

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Diarrhea-causing Escherichia coli (E. coli) possess colonization antigens or adhesins that enable the bacteria to colonize the small intestines [3]. The K99 (F5) fimbrial antigen has been reported to be associated with a majority of enterotoxigenic E. coli (ETEC) isolated from cases of diarrhea in neonatal calves [1,8].
In India, diarrhea in calves poses a major threat to the health of the animals and leads to economic loss. Little systematic work has been done to obviate this threat. One reason could be the absence of diagnostic tests to detect pathogenic E. coli strains. While there are some reports on human ETEC infections in India [4,9,10], very little information is available on ETEC-mediated diarrhea in neonatal calves [12].
A number of diagnostic tests are currently available for detecting ETEC. Double-antibody enzyme-linked immunosorbent assay (ELISA) was developed to detect the K99 pilus antigen [7]. DNA gene probes specific for genes encoding toxins and adhesins of ETEC [27] and multiplex polymerase chain reaction (PCR) for the rapid screening of ETEC toxins [24,26] have also been used with a fair amount of success. However, these tests require proper facilities and some amount of scientific expertise to conduct and interpret the test results. Therefore, we developed a simple but specific test to detect K99+ E. coli recovered from feces of diarrheic calves. The K99 fimbrial antigen was isolated and purified, monoclonal antibodies (MAbs) were produced against K99, and a co-agglutination test was developed to detect K99+ E. coli.
Materials and Methods
Bacteria, media and antibodies
E. coli were isolated from fecal samples collected from diarrheic calves. The isolates were grown in Minca-Isovitalex medium as described by Guinee et al. [6]; the medium was supplemented with 1 g of yeast extract (Oxoid) per liter of medium.
K99+ E. coli isolates were initially identified by agglutination tests using K99 antiserum obtained from the National Institute of Public Health and Environmental Protection (Netherlands), and subsequently confirmed by electron microscopy. K99 antigen was isolated and purified from a field isolate, designated SAR-14, which exhibited strong agglutination with K99 antiserum. The reference K99 (F5) MAb was procured from the Central Veterinary Laboratory (CVL), UK.
Electron microscopy
Electron microscopy was carried out as described by Korhonen et al. [11]. SAR-14, a wild strain of E. coli, was grown in Minca-Isovitalex media, prepared as described by Guinee et al. [6]; for 17 h, stained with 1% (w/v) phosphotungstic acid and examined in a electron microscope (JEM 100; Jeol, Japan) at an operating voltage of 80 kV.
Isolation of K99 antigen from a wild strain of E. coli
The SAR-14 strain of E. coli was grown in 3.0 l of Minca-Isovitalex broth for 17 h at 37℃ (O.D.660 = 1.6). The bacteria were then harvested by centrifugation at 6,000 g and resuspended in phosphate urea buffer (50 mM phosphate buffer, pH 7.2 with 2M urea) at O.D.660 = 100. The suspension was heated at 60℃ for 20 min and centrifuged at 30,000 × g for 15 min. The sediment was discarded, while the K99 antigen in the supernatant was precipitated with ammonium sulfate, separated and dialyzed as per Morris et al. [17].
Gel filtration chromatography
A glass column (Pharmacia, Sweden) measuring 60 cm in length by 1 cm in diameter was packed with Sepharose CL-4B (Pharmacia, Sweden) to a bed volume of 35 ml with a peristaltic pump. The packed column was washed with sodium phosphate buffer (50 mM, pH 7.2) and equilibrated with several column volumes of phosphate buffer containing 2M urea (PUB). The salt-precipitated bacterial proteins (in PUB) were gently loaded on the column and 60 fractions of 1-ml were collected.
Spectrophotometric readings of each fraction were taken at 280 nm. Fractions constituting individual peaks were pooled and analyzed for K99 antigen. Concentrated, pooled fractions were dialyzed for 72 h against phosphate-deoxycholate (DOC) buffer (phosphate buffer, pH 7.5 containing 0.5% sodium deoxycholate) after addition of DOC to the fraction [0.5% DOC (w/v)]. The purity of the fractions was checked by SDS-PAGE.
Fast protein liquid chromatography (FPLC)
The FPLC system (Amersham Pharmacia Biotech, USA) equipped with cation exchange column MonoS HR 5/5 was used for purification. The column was equilibrated in buffer A (10 mM phosphate buffer, pH 7.2), and bound proteins were eluted in buffer B (10 mM phosphate buffer containing 250 mM NaCl) with a phosphate buffer-NaCl gradient of 0-100%.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE)
SDS-PAGE gels were prepared by the Laemmli method [13] with the modification of Lugtenberg et al. [14]. Electrophoresis was carried out after loading 2 µg of sample per lane, along with a lane of standard molecular weight markers (10 kDa ladder; Gibco BRL, USA). Gels were stained with Coomassie Brilliant Blue.
Immunoblotting
Crude and purified protein fractions were subjected to Western blotting as described by Sambrook et al. [20]. Proteins were separated by electrophoresis on SDS-polyacrylamide gels and transferred to nitrocellulose membrane using LKB 2117 Electrophoresis unit, NOVABLOT (Pharmacia, Sweden). Membranes were incubated with blocking solution (1% skimmed milk powder in distilled water) for 2 h to avoid non-specific binding. The reference anti-K99 MAbs (CVL, UK) were diluted 1 : 500 and incubated with the membrane for ~2 h. After washing with Tris-Cl buffer, pH 7.5, rabbit anti-mouse IgG (diluted 1 : 1,000 with Tris-Cl buffer) conjugated with horseradish peroxidase (HRPO) was incubated with the membranes for 2 h at ambient temperature. The proteins were then stained with the HRPO substrate diaminobenzidine.
Dot immunoblots
Protein fractions eluted from the Sepharose CL-4B column were dotted on nitrocellulose membranes. The dot-blots were sequentially incubated with standard anti-K99 MAbs and rabbit anti-mouse IgG-HRPO conjugate and developed as described above.
Hyper-immunization of rabbit with K99 antigen
A healthy 12-week-old female New Zealand White rabbit was selected for raising polyclonal antibodies against purified K99 antigen. Pre-immune serum was collected and stored at -20℃ after addition of sodium azide (0.005%). About 100 µl of purified K99 antigen (~200 µg protein) was emulsified with 100 µl of Titermax (CytRx, USA) adjuvant and injected intramuscularly in both hind legs of the rabbit. Thereafter, injections of 50 µg purified K99 antigen in 100 µl of PBS pH 7.3 were given intravenously on days 31, 38 and 47.
Serum samples were collected on days 14, 21, 29, 44 and 55, and antibody titers were estimated by ELISA. The final bleed was collected on day 65, and the serum was preserved at -20℃.
Monoclonal antibody production
Hybridoma technology [15] was used to produce K99-specific MAbs. Briefly, purified K99 antigen (50 µg) was emulsified with Titermax adjuvant and injected intramuscularly in 8-week-old BALB/c mice twice with a 3-week interval. This procedure was followed by two intravenous inoculations of 20 µg and 30 µg of antigen, respectively, with a 3-day interval. The mice were regularly bled to assay antibody titers and, when sufficient titers had developed (~ 2 months), the mice were sacrificed and their spleens were removed aseptically. Hybridomas were produced by polyethylene glycol-mediated fusion of spleen cells with the mouse plasmacytoma cell line SP2O1-Ag-14. The resulting hybridomas were grown in selective medium containing hypoxanthine, aminopterin and thymidine in 24-well tissue culture plates. Supernatants from wells containing hybridoma clones were assayed for K99-reactive antibodies by ELISA using purified K99 as the antigen. A specific clone, designated NEK99-5.6.12, was selected and propagated in large tissue-culture flasks. The secreted antibodies were harvested by centrifugation at 5,000 × g, precipitated with ammonium sulfate (45% saturation) and dialyzed against phosphate buffer, pH 7.2. The antibodies were affinity-purified on a Protein G Sepharose column (Pierce, USA.) and subsequently characterized by ELISA using the mouse monoclonal antibody isotyping kit (ISO-2; Sigma, USA).
ELISA for screening of hybridoma culture supernatants
Polystyrene microtiter plates (Maxisorp; Nunc, Denmark) were coated with 100 ng/well of purified K99 antigen and blocked with 200 µl of 1% skimmed milk powder. Tissue culture supernatants were added to individual wells and the plates incubated for 2 h at 37℃. After washing, 100 µl of HRPO-conjugated rabbit anti-mouse IgG (diluted 1 : 1000) was added. The plates were incubated for 2 h at 37℃, then washed, and the substrate, tetramethyl benzidine with H2O2 (Genei, India) diluted 1 : 20 in distilled water, was added. The reaction was terminated with 50 µl 1 N H2SO4, and the plates were read in an ELISA reader (Titertek Multiskan; Titertek, Finland) at 490 nm. Standard monoclonal antibodies were used as positive controls and growth medium was used as the negative control.
Antigen capture ELISA
Microtiter plates were coated with an optimal concentration (~200 ng/well) of affinity-purified IgG from sera of rabbits hyper-immunized with purified K99 antigen. Adequate concentration (~2 × 105 cells/well) of bacterial cells from different isolates were added to duplicate wells and incubated for 2 h at 37℃. The wells were then washed with Tris-Cl buffer, pH 7.5, and standard monoclonal antibodies (CVL, UK) or test monoclonal antibodies (tissue culture-derived) were added to each of the duplicate wells, respectively. After incubation and washing, rabbit anti-mouse-HRPO conjugate was added, and the reactions were developed by substrate addition as described above. A standard E. coli strain positive for K99 expression (ATCC 31616) was used as positive control, and the laboratory E. coli strain HB101 was used as negative control.
The co-agglutination test
The adopted protocol was that described by Batra et al. [2], which was developed to detect Brucella antigens. Briefly, cells were harvested from overnight cultures of Staphylococcus aureus (S. aureus) Cowan I strain, washed in 0.01 M PBS pH 7.2 and resuspended at 5% (v/v) in the same buffer. Cells were sequentially treated with formalin (2% v/v, overnight at 4℃) and heat (80℃ for 5 min) and then stained with Giemsa for 6 h at 4℃. Stained cells were again washed and adjusted to 5% v/v in PBS. The cells were coated with purified reference or tissue culture-derived monoclonal antibodies at varying concentrations (0.5 to 5 mg), incubated at 37℃ for 2 h and washed to remove unbound antibodies. Aliquots of the monoclonal antibody-coated S. aureus were kept at 4℃ and at room temperature. Ten microliters of this suspension was mixed with 10 µl of bacterial suspension derived from feces of diarrheic calves on a test card. Agglutination appearing within a minute was considered a positive reaction. Positive and negative controls were the same as those used in antigen capture ELISA.
For hybridoma work, mice experiments were conducted according to the norms of "Committee for the Purpose of Control and Supervision of Experiments on Animals," Government of India, Ministry of Environment & Forests, Animal Welfare Division. Approval was obtained from the Institutional Animal Ethics Committee under Project Proposal No. 12/Departmental Proposal No. AH11.
Results
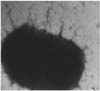
Following growth in Minca-Isovitalex broth for 17 h, fimbriae on E. coli strain SAR-14 were observed by electron microscopy (Fig. 1). The fimbriae exhibited agglutination with standard reference K99 monoclonal antibody. When grown in Nutrient agar or MacConkey agar, the SAR-14 strain did not agglutinate with K99 antiserum. Therefore, Minca-Isovitalex medium was appropriate for E. coli fimbrial K99 antigen expression.
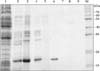
Heat treatment of fimbriated E. coli for 20 min at 60℃ in the presence of 2M urea effectively detached the fimbriae from the host cells. Detachment was verified by SDS-PAGE; a prominent 18.5-kDa band (the putative K99 antigen) was observed along with several minor higher molecular weight proteins in the supernatants of bacterial heat extracts (Fig. 2). The 18.5-kDa band was absent from the pellet fraction (Fig. 2). Proteins in the supernatants were further concentrated by ammonium sulfate precipitation (60% saturation), which appeared to enrich the putative K99 antigen (Fig. 2).
The heat-extracted, salt-precipitated fimbrial antigens were fractionated on a Sepharose CL-4B column equilibrated with PBS, pH 7.2, containing 2M urea. The proteins eluted in one major and one minor peaks (Fig. 3). Fractions comprising the two peaks were pooled separately and aliquots of the two fractions were tested by immunoblotting using reference MAbs to the K99 antigen. Dot blots confirmed the presence of K99 antigen in the heat extract, salt precipitate and the first sepharose gel-eluted protein peak (Fig. 3). The column-eluted fractions were analyzed on SDS polyacrylamide gel, which indicated that the first peak contained the 18.5-kDa protein along with several minor proteins of moderate sizes. The second peak contained minor amounts of high molecular weight proteins (Fig. 2). The 18.5-kDa protein was further purified by treatment with sodium deoxycholate, which appeared to specifically solubilize the 18.5 kDa protein (Fig. 2); minute amounts of a ~38-kDa protein also appeared to co-solubilize with the 18.5-kDa protein (Fig. 2). Complete purification of the desired protein, from the major peak of sepharose gel-eluted proteins, was achieved by FPLC using the cation-exchange column MonoS HR 5/5. Two sharp peaks were observed on FPLC analysis (Fig. 4). The first peak represented unbound proteins, which appeared in the flow-through fraction. The second peak represented bound proteins that eluted with increasing concentrations of NaCl in the buffer. Dot-blot analysis using reference K99 MAbs confirmed that only the second peak contained K99 antigen (data not shown). Finally, various fractions obtained throughout the purification procedure were analyzed by Western blotting using reference K99 MAbs. The single band migrating at ~18.5 kDa was confirmed as purified K99 antigen (Fig. 5).
Purified K99 protein was used to immunize BALB/c mice for MAb production. Actively growing hybrids were seen in the microtiter plates a week after fusion of mouse spleen cells with myeloma cells. Supernatants from these hybrids were collected and assayed for K99 reactivity by ELISA. Two stable hybrids (Nos. 5 and 3) that produced large amounts of K99-specific antibodies were selected and cloned by limited dilution. Clones 5.6 and 3.8 were subcloned twice and the final clones, designated NEK99-5.6.12 and NEK99-3.8.1, respectively, were cryopreserved in liquid nitrogen. Antibodies derived from these two clones were compared with the reference MAb (CVL, UK) in Western blot analysis of proteins eluted from the gel filtration column and FPLC-purified protein. The reactivity of both the tissue culture-derived antibodies (5.6.12 and 3.8.1) was comparable to that of the reference MAb (CVL, UK) (Fig. 6). Clone NEK99-5.6.12 was selected for further applications. The specificity of antibodies derived from this clone was checked at each stage of subcloning and when cells were revived after cryopreservation.
The tissue culture-derived MAbs were identified as IgG1 subclass by standard isotype ELISA. The reference antibody, known to have IgG2a specificity, gave the expected results and showed maximum reactivity to IgG2a subclass (data not shown).
Tissue culture-derived MAbs were purified by affinity chromatography using protein G columns (Amersham Biosciences, UK). The purified MAbs were tested for cross reactivity to different Gram negative bacteria in Western blot analyses. Whole cell lysates of E. coli (SAR-14), Pasteurella, Enterobacter, Pseudomonas and Klebsiella were separated by SDS-PAGE, and the proteins were transferred to a nitrocellulose membrane. Tissue culture-derived MAbs were allowed to bind to the membrane. The antibody specifically reacted with the K99 antigen of E. coli and did not bind to proteins from other Gram negative bacteria, with the exception of a ~20 kDa protein in Klebsiella (Fig. 7). Despite the cross-reactivity with Klebsiella antigen, these bacteria are easily differentiable on classical media.
Various concentrations of purified monoclonal antibodies from clone NEK99-5.6.12 were used to coat the S. aureus Cowan I strain. We found that 0.5 µg of antibodies per mL of S. aureus suspension was adequate to elicit a positive reaction with the standard K99+ E. coli strain in a co-agglutination test, reflecting the high sensitivity of the assay. Antibody-coated S. aureus were stable for 3 months when stored at ambient temperature (20℃ to 39℃) and for more than 5 months when stored at 4℃.
The co-agglutination test was used in testing field isolates for the presence of K99+ E. coli. The specificity of the tissue culture-derived antibodies was also compared to that of the reference monoclonal antibody when the two were separately coated on colored Staphylococcus cells and tested for agglutination of random field isolates. Both antibodies exhibited similar reactions for all 25 strains tested, except for BOM-11.2/95, which reacted positively with the tissue culture-derived MAb but did not react with the reference MAb (Table 1).
Monoclonal antibodies prepared in the laboratory were used in a double antibody sandwich ELISA to assess their ability to identify K99+ E. coli present in fecal specimens (Table 2). The results were compared with those obtained in the co-agglutination test. A higher absorbance value (OD450 > 0.7) was generally associated with a positive reaction in co-agglutination test. However, two strains (KUS-5.1/95 and BOM-17/95) with absorbance values of 0.74 were negative in the co-agglutination test; two others (BOM-5.1/95 and BOM-59.1/95) had absorbance values of less than 0.7 but were positive in the co-agglutination test.
Discussion
Escherichia coli is a member of the normal gut flora in humans and animals and its presence may have nutritional significance [21]. However, it is difficult to distinguish these bacteria from pathogenic E. coli using routine bacteriological methods like light microscopy, colony morphology, biochemical characteristics, etc. Still, it is important to demonstrate the presence of pathogenic E. coli before attributing it as a cause of colibacillosis. Virulence factors associated with enterotoxigenic E. coli in calves have been characterized, and the K99 antigen has been used to develop sensitive tests for diagnosis or subunit vaccines [21].
Several procedures are available to obtain purified bacterial fimbriae [1,22]. The major problem encountered with these procedures is that isolated fimbriae may be contaminated with outer membrane proteins, which are difficult to remove due to their hydrophobic binding to fimbriae. Gel filtration chromatography alone resulted into only partial fractionation. Peak 1 contained many contaminating proteins along with the K99 antigen. Treatment with DOC, which acts similarly to Triton X-100, appeared to specifically solubilize the K99 protein and not the outer membrane proteins. However, minute amounts of a ~38-kDa protein co-solubilized with the K99 antigen. This was confirmed on Western blots, where this higher molecular weight (Mw) protein reacted with three different MAbs to the K99 antigen. This suggested that the high Mw protein is either a multimeric form of K99 protein or is another fimbrial protein having shared epitopes with the K99 antigen. Following FPLC purification, the high Mw protein was not observed in SDS gels, implying that the former is more likely . The cation exchange column was used because K99 protein is reported to have a pI value of > 9 [1]. A combination of gel filtration and ion-exchange chromatography, rather than detergent solubilization, provided suitable, quicker, reproducible and complete purification of the K99 antigen.
The purified K99 antigen was primarily composed of protein subunits with an apparent molecular weight of 18,500 Daltons based on SDS polyacrylamide gel analysis. There are variable reports regarding the molecular weight of K99 antigen. de Graaf et al. [5] and Vazquez et al. [25] found the molecular weight of K99 antigen to be 18,500 and 17,000 respectively. The latter report also suggests that a single fimbrial subunit may occur in different conformations and, thus, slightly different molecular weights may be found upon SDS-PAGE analysis [25]. In the current work, however, the presence of a single band with an apparent molecular weight of 18.5 kDa on SDS-PAGE indicated the purity of protein subunits; this protein was confirmed to be K99 antigen by immunoblotting using MAbs for K99 antigen. The positive control used in both tests was E. coli ATCC strain 31616 (K99+) and the negative control was HB101 (K99-).
The MAb to K99 antigen was tested for cross reactivity against whole cell lysates prepared from other Gram negative bacteria. All Gram negative bacteria were non-reactive to the MAb except Klebsiella. Cross reactivity amongst different subunits of the fimbrial protein is known to occur [19]. The fact that a MAb raised against E. coli fimbrial protein cross-reacted with Klebsiella suggests that there may be some common epitope(s) between the two. Further research on fimbrial proteins can explain this observation.
There are many advantages to preparing monoclonal antibodies to K99 antigen. Apart from economic considerations and the regular availability in adequate quantities, these antibodies could be orally administered to neonatal calves to reduce the economic losses of cattle owners during outbreaks of enteric colibacillosis in unvaccinated herds. Production of K99-specific antibody and development of sensitive ELISAs to detect K99 fimbriae on ETEC cells were reported earlier [5, 16]. Co-agglutination tests using Staphylococcus aureus have been used to diagnose H. pleuropneumonia infections in pigs, pneumococcal pneumonia in humans and brucellosis in cattle [2]. MAbs also have important applications in the detection of K99 adhesin of E. coli in aqueous vaccines [23]. In the present work, monoclonal antibodies raised against K99 antigen used in a Staphylococci agglutination test specifically detected K99+ E. coli in fecal isolates from diarrheic calves. Antigen capture ELISA confirmed the specificity of the co-agglutination test. A combination of polyclonal and monoclonal antibodies was used in the antigen capture ELISA; polyclonal antibodies were used as capture antibodies and monoclonal antibodies were used as detector antibodies, as suggested by Raybould et al. [18].
Staphylococci coated separately with tissue culture-derived and reference K99 MAbs reacted similarly in co-agglutination tests against various field isolates, except against the isolate, BOM-11.2/95. It is not clear whether there is subclass specificity between the 2 MAbs. The MAb we developed is IgG1, whereas the reference MAb is IgG2a.
The co-agglutination test that we developed provides convenient, simple and rapid screening of fecal isolates for presence of K99+ E. coli. It does not require expensive facilities and can therefore be used cost-effectively. This test could have important applications in epidemiological studies where the incidence of enterotoxigenic E. coli in a particular region can be studied. Based on incidence rates, decisions could then be made for the development and use of a vaccine to prevent and control colibacillosis in calves.







 XML Download
XML Download