

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Since it was first discovered in California in 1981, AIDS has been considered one of the worst diseases of mankind that has been rapidly increasing around the world [7]. Anti-human immunodeficiency virus (HIV) drugs can be divided into several classes depending on their mechanism of action, including the interception of the HIV inflow into immunocytes, the inhibition of reproduction and proliferation within cells, and the suppression of viral release from the cells after reproduction. The development of non-nucleoside reverse transcriptase inhibitors has also received much attention as another class of HIV genome replication inhibitors [2,3,14]. However, this group of drugs often shows structural hydrophobicity, and low solubility resulting in poor oral bioavailability. For these reasons, there have been several attempts to improve their oral bioavailability. We recently developed a non-nucleoside reverse transcriptase inhibitor 1-(2-amino-pyridin-4-ylmethyl)-6-(3,5-dimethyl-benzoyl)-5-isopropyl-1H-pyrimidine-2,4-dione (VP-0502) that is a derivative of VP-0501 [4]. This compound however has a very poor absorption profile after oral administration. For this reason, we designed an amino acid-attached prodrug of VP-0502 by binding the amino acid alanine to improve its oral bioavailability. This paper describes a pharmacokinetic comparison between VP-0502 and its prodrug alanine amide of VP-0502 (VP-0502AL). This could lead to the development of an oral drug delivery system from the intestine.
Materials and Methods
Chemicals and animals
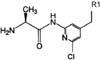
1-(2-amino-pyridin-4-ylmethyl)-6-(3,5-dimethyl-benzoyl)-5-isopropyl-1H-pyrimidine-2,4-dione (VP-0502) and alanine amide of VP-0502 (VP-0502AL) (Fig. 1, purity >96%) were investigated in this study. Healthy, male rats (Sprague-Dawley) of approximately 8 weeks old and weighing from 220 to 280 g (Orient, Korea) were used in this experiment. The animals were determined to be healthy on the basis of clinical examination. They were held under semi-barrier conditions at a constant 12 h cycle of light and dark, were fed on a laboratory rodent diet (Pico 5053; Labdiet, Korea) and had free access to drinking water throughout the experimental period.
Drug preparation and administration
Animals were fasted for approximately 12 h prior to dosing. For the intravenous bolus study, VP-0502 and VP-0502AL were dissolved in 40% polyethylene glycol 400 (Fluka, USA) and injected via the tail vein at a dosage of 20 mg/5 ml/kg b.w. For the oral study, VP-0502 and VP-0502AL were suspended in 14.3% citric acid (Duksan, Korea) which was administered at a dosage of 100 mg/10 ml/kg. A syringe fitted with a stomach tube was used for the oral administration. Each animal was weighed just before drug treatment to ensure that the precise dose was given.
Blood sampling
200 µl blood samples were collected from the tail vein using a vacutainer (Becton-Dickinson, USA) into a plastic tube containing EDTA as an anticoagulant (final concentration 0.2%). Serial samples were obtained at the following times after dosing: (i) In the intravenous study, pre-dosing (0), 0.5, 1, 2, 4, 6 and 8 h. (ii) In the oral study, pre-dosing, 0.5, 1, 2, 3, 4, 5, 6, 7 and 8 h. Immediately after collection, the blood samples were gently inverted and placed on ice. The samples were then centrifuged at 16,700 g (Hanil Microspin, Korea) for 5 min and the plasma was recovered and maintained frozen at -20℃ until analyzed within 2 weeks of sampling. Frozen plasma samples were thawed at room temperature prior to extraction, and each aliquot was mixed with three volumes of methanol and vortexed (Thermolyne, USA). The resulting mixture was centrifuged at 12,000 rpm for 2 min, and the supernatant was injected directly onto the high-performance liquid chromatography (HPLC) column.
Drug analysis
A microbore HPLC system including a photodiode array UV detector (Agilent, USA), a pump (Agilent, USA), a degasser (Agilent, USA), an autosampler (Agilent, USA), and a column oven (Agilent, USA) was used to measure both VP-0502 and VP-0502AL in rat plasma. The analytical column was a Capcell-pak C18 column (UG120, 100 × 2.0 mm I.D., 5 µm; Shiseido, Japan). The optimal mobile phase conditions for both analytes were selected using an acetonitrile- or methanol-water gradient system and the UV wavelength was set at 264 nm. The mobile phase was a mixture of 0.01 M dibasic potassium phosphate (Sigma, USA) and acetonitrile (60 : 40, v/v). The flow rate was 0.2 ml/min and the column-oven temperature was set at 25℃. The injection volume was 5 µl. The stability of the compounds in rat plasma was also studied over two weeks of sampling.
Pharmacokinetics
The pharmacokinetic parameters were obtained using a pharmacokinetic program "WinNonlin", fitting data to a one-compartment open model. For the intravenous injection, the pharmacokinetic parameters including elimination rate constant (kel), distribution volume (Vd), elimination half-life (t1/2), concentration right after administration (C0), total body clearance (CLtot), area under the concentration-time curve (AUC), area under the moment curve (AUMC), and the mean residence time (MRT) were estimated from the model interpretation. The formula for the concentration in plasma for intravenous injection was C(t) = D/Vd · exp-kel · t, where C is the plasma concentration and D is the dose. For the oral administration study, the lag time was considered and the absorption and elimination rates were applied as first-order kinetics. Model-dependent parameters including the lag time (tlag), absorption rate constant (ka) and kel, absorption half-time (t1/2,a) and t1/2, the time to reach the maximum concentration (tmax), maximum concentration (Cmax), and AUC were calculated. Bioavailability (F) was calculated from the equation of F(%) = (AUCpo/AUCiv) · (Div/Dpo). The formula for oral administration was C(t) = D · ka/Vd/(ka-kel) · (exp-kel · t-exp-ka · t). Plasma drug concentrations and the estimated pharmacokinetic parameters were reported as mean ± SD (n = >3).
Results
Clinical observation
No particular clinical findings were observed after administration of VP-0502 and VP-0502AL by either route to rats.
Drug analysis
The chromatograms of both analytes, VP-0502 and VP-0502AL, are shown in Fig. 2. The UV wavelength was set at 264 nm based on the λmax value from the result of UV spectra scanning. Under these conditions, VP-0502 and VP-0502AL were separated well on the chromatogram and were eluted within 10 min. The recoveries of VP-0502 and VP-0502AL were 77% and 87%, respectively. The limits of quantification (LOQ) were lower than 50 ng/ml for both analytes.
Pharmacokinetics
VP-0502 was not detected in the plasma following oral administration to rats at a dose rate of 100 mg/kg. However, it was readily found in the plasma following intravenous injection at a dose of 20 mg/kg, though the plasma concentration level was minimal (Fig. 3). Table 1 shows the pharmacokinetic parameters of VP-0502 injected intravenously, where the AUC, t1/2, C0, CLtot, AUMC, and MRT were 0.3 µg · h/ml, 1.6 h, 0.2 µg/ml, 69 l/h/kg, 0.7 µg · h2/ml and 2 h, respectively.
When VP-0502AL was orally administered, both VP-0502 and VP-0502AL were quantitatively observed in plasma as shown in Fig. 3. The pharmacokinetic parameters of VP-0502AL including tlag, tmax, Cmax, t1/2, F and AUC were 1.6 h, 2.7 h, 0.2 µg/ml, 1 h, 3.4% and 0.5 µg · h/ml, respectively (Table 2). Moreover, the active metabolite (VP-0502) was found in the plasma at significantly higher levels than the parent compound (VP-0502AL) with Cmax of 0.8 µg/ml and AUC of 2 µg · h/ml (Fig. 4). When VP-0502AL was intravenously injected, both VP-0502 and VP-0502AL appeared extensively in plasma as shown in Fig. 5. The pharmacokinetics parameters of VP-0502AL were Vd: 5 l/kg, AUC: 3 µg · h/ml, t1/2: 0.5 h, C0: 6 µg/ml, CLtot: 7 l/h/kg, and AUMC: 2 µg · h2/ml (Table 3).
Discussion
We performed a pharmacokinetic study of VP-0502 and its prodrug VP-0502AL after oral and intravenous administration to rats. No particular clinical changes were observed in any of the treated animals. This indicates that a single dose (100 mg/kg orally or 20 mg/kg intravenously) of either drug did not generate any visible clinical side effects in rats.
For the determination of VP-0502 and VP-0502AL in the plasma, we developed an analytical HPLC system using photodiode-array detection. Both VP-0502 and VP-0502AL were successfully separated on the chromatogram within a 10 min retention time with no interceptive peak at the selected 264 nm wavelengths. For the mobile phase, the optimal conditions were obtained from a mixture of phosphate buffer solution (K2HPO4, pH 7) and acetonitrile (60 : 40, v/v). The ODS column was selected over the others (C18, C8 and phenyl bonded), because it revealed excellent resolution properties. The LOQs and recoveries were found to be sufficient to detect both analytes in the plasma samples. Therefore, the current HPLC system is considered an appropriate technique for simple and rapid determination of the VP-compounds for pharmacokinetic analyses in rats.
Following oral administration of VP-0502, the analyte was not detectable in the plasma, but it was readily detected following the intravenous injection. This indicates that the bioavailability of VP-0502 is negligible and thus its application in practice has no significance in terms of its poor oral pharmacokinetic profiles. It is therefore necessary to develop a drug delivery system (DDS) in order to improve the bioavailability of VP-0502. Accordingly, an amino acid formula (pro-drug VP-0502AL) was designed for this study. This type of amino acid-attached DDS not only increases the solubility but also it improves its pharmacokinetic profile [1]. For instance, when L-glutamic acid was added to the formula, the receptivity went up and the drug could be effectively delivered to a target [15]. Furthermore, many research works attempt to improve the oral absorption of poor-soluble drugs by increasing the solubility for which amino acid attachment is considered. Thus, structures have been developed that increase the receptivity via combination with one to three amino acids, enable the transformation of the pro-drug to active metabolite, which targets the hydrolases of digestive mucosa, [5,6,8-13,16].
Although VP-0502 was not detected in plasma when administered orally, both the parent compound VP-0502AL and its active metabolite VP-0502, simultaneously appeared in the plasma following the oral administration of VP-0502AL, with significant Cmax of 0.8 µg/ml and an AUC of 2 µg · h/ml. Furthermore, the VP-compounds were maintained in the plasma for over 7 h. This result shows that the bioavailability of VP-0502 when administered as an amino acid-attached pro-drug was notably increased. Data collected after oral administration of VP-0502AL were best fitted to a one-compartment open model with the first-order output. The approximate time to reach the maximum concentration was about 3 h and the maximum plasma concentration was 0.2 µg/ml. The elimination half-life was about 1 h, and the area under the plasma concentration-time curve of 0.5 µg · h/ml was sufficiently to suggest the efficiency of the prodrug, compared to VP-0502. Additionally, in terms of pharmacokinetics, rapid oral absorption and transformation to an active form, and the maintenance of concentration in plasma were satisfactory for a new prodrug candidate. The improvement of bioavailability by the amino acid alanine formula for VP-0501AL performed prior to this, has shown a similar result to this test [4]. This confirmed that amino acid alanine attachment is very effective approach to making a prodrug. Therefore, VP-0502AL is expected to become a highly bioavailable anti-AIDS drug candidate and/or lead compound.







 XML Download
XML Download