

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Bovine spongiform encephalopathy (BSE) is thought to be among the transmissible spongiform encephalopathies (TSEs), also referred to as prion diseases, which are fatal neurodegenerative disorders and share important mechanistic aspects with other, more frequently occurring diseases such as Alzheimer's, Huntington's, and Parkinson's disease [11,26]. The TSEs, including BSE, are characterized by neuronal vacuolation and accumulation of the abnormal isoform (PrPSc) of a host-encoded cellular membrane glycoprotein, referred to as normal prion protein (PrPC), in the central nerve system [14,38]. BSE, which reached epidemic proportions in Britain in the 1990s and is increasing in many other countries as well, has been transmitted to more than 100 human beings through the consumption of infected beef [9,11]. Due to the risks to public health as a causative agent of variant Creutzfeldt-Jakob disease (vCJD) and its role as an example of a novel mechanism of biological information transfer based on the transmission of protein conformation rather than on the inheritance of a nucleic acid sequence, vCJD has now become a subject of general interest [1,9,27,36]. In countries from which the emergence of BSE has not yet been reported, its risks to cattle and public health still remain, and an active screening method and means of control for BSE are strongly needed.
Beginning with research on the pathogenesis of BSE, lots of efforts have been focused on topics such as the physiological roles of PrPC, as well as transmission, diagnosis, therapy, and prophylaxis. The normal function of PrPC remains to be established. However, its localization on the cell surface via a glycosylphosphatidylinositol (GPI) anchor would be consistent with roles in cell adhesion and recognition, ligand uptake, or transmembrane signaling, as well as neuroprotective function [4,5,7,8,11,25,26]. The conformational conversion of PrPC into the abnormal isoform PrPSc, as well as depletions, mutations, or topological aberrations in prion protein gene (Prnp), may lead to loss-of-function components in prion disease [9,22,23,30]. Despite its potential risk as a zoonotic disease, the proteinase K-resistant property of PrPSc is the only pathway for diagnosis. Currently, the main method of diagnosis of BSE is based on the postmortem detection of PrPSc by means of immunological techniques such as enzyme-linked immunosorbent assay, Western blot, and immunohistochemistry, together with histopathology [12].
Chinese hamster ovary (CHO-K1) cells have recently been used to study gene expression, toxicity screening, cell biology, and virology, as well as prion disease [1,3,6,19,33]. A detailed analysis of the characterization of CHO-K1 cells transfected with a Prnp has not been performed, in spite of the fact that it is an essential component of the cellular biology of prion disease. Moreover, most BSE research has been deduced from experimental mouse Prnp models, in vitro and in vivo, since these models are well-understood and easy to use even though the nucleotide and predicted amino acid sequence of the bovine Prnp ORF are approximately 78% and 84% homologous to those of the mouse, respectively [15].
To better understand and control BSE, a BSE-specific experimental model is needed. As basic research of BSE biology, we attempted to constitute the immortalized cell line stably expressing bovine Prnp with CHO-K1 cells and investigate the characteristics, which followed the gene expression using different parameters of cell biology.
Materials and Methods
Cell culture of CHO-K1 cells
The wild-type CHO-K1 cells were purchased from the Korean Cell Line Bank (KCLB No. 10061) and maintained in Dulbecco's modified Eagle's medium (DMEM; Gibco, USA) with a high glucose concentration (4.5 g/l) supplemented with 10% fetal bovine serum and 2 mM glutamine, penicillin, and streptomycin at 37 in a 5% CO2 incubator.
Transfection of bovine Prnp
The cDNA encoding the Prnp ORF of Korean cattle, with 264 amino acids and a predicted molecular weight of 28 kDa [15], was cloned into the pIRESpuro2 eukaryotic expression vector (Clontech, USA), and was transfected in the CHO-K1 cells using lipofectamine (Invitrogen, USA). The CHO-K1 cells expressing bovine prion protein were constructed by selection of the puromycin-resistant cells in the complete medium containing 30 µg/ml puromycin. To obtain one cell clone, the puromycin-resistant cells were resuspended and the dilution was calculated to give 2 cells/ml in the complete medium. The diluted cells were plated into 96-well plates and incubated for a week at 37 under 5% CO2.
PCR and RT-PCR
The insertion and expression of the cloned cDNA were analyzed by genomic PCR and RT-PCR, respectively. Genomic DNA from each clone was extracted using a genomic DNA purification kit (Promega, USA), and PCR was performed to screen the gene transfer with a forward primer, 5'-GAATTCATGGTGAAAAGCCACATAGGCAGTTGG-3', and a reverse primer, 5'-GAATTCCTATCCTACTATGAGAAAAATGAGGAA-3'. The PCR amplification consisted of an initial denaturation at 94℃ for 5 min, followed by 30 cycles of denaturation at 94℃ for 30 sec, annealing at 60℃ for 30 sec, extension at 72℃ for 1 min 30 sec, and then a final extension at 72℃ for 15 min. PCR products were analyzed by electrophoresis on 1.0% agarose gel. Total RNA was isolated from the positive one cell clones using Trizol reagent (Invitrogen, USA) and chloroform. Aqueous total RNA was precipitated by the addition of isopropanol and centrifuged at 12,000 × g for 10 min. The RNA pellets were washed once with 75% ethanol and dissolved in diethylpyrocarbonate-treated water. The RNA was treated with 2 units of RNase-free DNase at 37℃ for 30 min to remove the residual DNA. Single-stranded cDNA was synthesized using the superscript III preamplification system for the first-strand cDNA synthesis system (Invitrogen, USA). Five µg of purified total RNA was incubated with 100 units of superscript III reverse transcriptase at 50℃ for 50 min in the presence of 10X RT buffer, 25 mM MgCl2, 0.1M DTT, 10 mM dNTP, and 50 µM oligo dT. The synthesized single-stranded cDNA were treated with 2 units of RNase H at 37℃ for 20 min to remove RNA, and PCR was then performed as described above to screen for gene transcription of bovine Prnp.
Immunoprecipitation
Cells were lysed with cold RIPA buffer (50 mM Tris-HCl, 150mM NaCl, 1mM PMSF, 1mM EDTA, 5 µg/ml aprotinin, 5 µg/ml leupeptin, 1% TritonX-100, 1% sodium deoxycholate, 0.1% SDS), and the lysate was precleared by the addition of control mouse IgG purified using HiTrap protein G HP (Amersham, Australia), together with protein G agarose (Santa Cruz, USA). The supernatant was then incubated with 6H4 monoclonal antibody, which was kindly provided by Dr. A. Zurbriggen [18]. Incubation was followed by the addition of protein G-agarose at 4℃; the mixture was subjected to soft shaking overnight. The immunoprecipitates were boiled with electrophoresis sample buffer (50 mM Tris-Cl, pH 6.8, 100 mM dithiothreitol, 2% SDS, 0.1% bromophenol blue, 10% glycerol) and loaded for 12% SDS-PAGE. Proteins were then transferred to nitrocellulose membranes (BioRad, USA) using a semidry blotting system. The blots were incubated with the 44B1 monoclonal antibody [17], and then with alkaline phosphatase-conjugated anti-mouse IgG. Detection was performed by visualization using BCIP/NTB substrate (BioRad, USA).
MTT and LDH assay
CHO-K1 cells and bovine Prnp-transfectants were plated in 96-well microplates at a density of 8 × 103 cells/well and cultured for 72 h at 37℃ under 5% CO2. For the MTT assay, the culture medium was replaced by 200 µl of fresh medium, and sterile filtered 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT; Sigma, USA) was added to each well, reaching a final concentration of 1.0mg/ml. Unreacted dye was removed after 4 h, the insoluble formazan crystals were dissolved in 200 µl of dimethylsulfoxide (Sigma, USA), and absorbance was measured at a wavelength of 570 nm. The relative cell proliferation (%) was related to a 100% confluence per well in the A570 test/A570 100% confluence well [24]. For the lactate dehydrogenase (LDH) assay, the concentration in the culture medium was measured using the homogeneous membrane integrity assay (CytoTox-ONE; Promega, USA), allowing the spectrophotometric determination of the nicotinamide adenine dinucleotide reduction at 490 nm. Controls were performed with 1% TritonX-100 and set as 100% LDH release. The relative LDH release (%) is defined as the ratio of LDH released to the total LDH in the intact cells.
NO production and SOD activity assay
CHO-K1 cells and bovine Prnp-transfectants were plated in 60 mm cell culture dishes at a density of 5.0 × 105 cells/dish. These cells were stimulated with 1 µg/ml of lipopolysaccharide (BioWhittaker, USA) or 5 µg/ml of concanavalin A (ConA; Sigma, USA) in serum-free medium (Opti-MEMI; Invitrogen, USA). The nitric oxide (NO) level from each culture supernatant was determined by the Griess reaction, using Griess reagent (1% sulfanilamide and 0.1% N-1-napthylethylenediamine dihydrochloride in 2.5% phosphoric acid). The culture media and serially-diluted sodium nitrite were used as standard references. Absorbances were measured with a 540 nm filter, and the concentration of NO was determined by comparison to the standard curve. Superoxide dismutase (SOD) activity was determined by using the superoxide dismutase assay kit (Cayman, USA) according to the protocol of the manufacturer, which utilizes a tetrazolium salt for the detection of superoxide radicals generated by xanthine oxidase. The stimulated cells from each time point were lysed and mixed with the radical detector, and the catalysis of SOD was then initiated by the addition of xanthine oxidase. The absorbances were read at 450 nm, and SOD activities of the samples were calculated using a serially-diluted SOD standard reference curve. SOD activity (unit/ml) was represented in units. One unit was defined as the amount of enzyme needed to exhibit 50% dismutation of the superoxide radical per milliliter.
DNA fragmentation and caspase-3 activity assay
CHO-K1 and bovine Prnp-transfectant were plated in 60 mm cell culture dishes at a density of 5.0 × 105 cells/dish, and were treated with 200 µg/ml cyclohexamide. For the DNA fragmentation assay, cells were collected at specific time points and lysed. Genomic DNA from the cell lysate was isolated, and the pattern of fragmented DNA was then analyzed by 1.5% agarose gel electrophoresis. Caspase-3 activity was measured by using assay system (CaspACE; Promega, USA) according to the protocol of the manufacturer. The cells from each time point were lysed, and the lysates were then reacted with the colorimetric substrate (Ac-DEVD-pNA) provided in the kit. The chromophore p-nitroaniline (pNA) released by caspase-3 from the cell lysate was monitored by a spectrophotometer at 405 nm. The activities of caspase-3 were calculated in comparison to the pNA calibration curve.
Results
Transfection of bovine Prnp
The pIRESpuro2 eukaryotic expression vector, which harbored cDNA encoding bovine Prnp ORF with 264 amino acids, was transfected into CHO-K1 cells, and the bovine Prnp-transfectants were selected by culturing in a complete medium containing puromycin. No specific morphological changes of transfected cells were detected by microscopic comparison with wild-type cells.
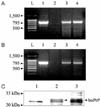
After drug selection, a limiting dilution was performed, and several clones of bovine Prnp-transfectant were obtained. Genomic DNA was analyzed by PCR in order to screen for the harboring of the transfected genes in each cell clone, and RT-PCR was then carried out to screen for gene transcription against the positive clones in the PCR analysis. The amplicons that were 795 bp in size, a size that is identical to that of full-length bovine Prnp ORF, were amplified by both PCR and RT-PCR; however, no specific band was observed in the lane of the CHO-K1 cells control (Fig. 1). Two clonal lines that expressed stable levels of bovine PrPC were obtained by screening gene transfer and transcription.
The expression of exogenous bovine prion protein was confirmed by Western blot analysis. Before Western blotting, prion proteins in transfected cells were immunoprecipitated by a combination of protein G-agarose beads and 6H4 monoclonal antibodies, which encompass amino acid residues 144 to 152. Two major bands with a molecular weight of about 31 kDa were detected (Fig. 1).
Biological characteristics of bovine Prnp-transfectant
Cell proliferation and viability rates of wild-type CHO-K1 cells and bovine Prnp-transfectants were measured by the MTT test and LDH assay. In the MTT test, wild-type cells showed a higher rate of proliferation (91.3 ± 2.4%) than the transfectant (68.7 ± 6.7%) (p < 0.05). In the LDH assay for cell viability, the relative LDH release of wild-type cells (2.7 ± 0.3%) was less than that of the transfectant (6.3 ± 0.9%) (p < 0.05) (Fig. 2).
NO and SOD Assay
In the NO assay, no detectable amount of nitrite was measured in the culture supernatant following stimulation with either LPS or ConA. The SOD activity in bovine Prnp-transfectant (5.2 units) was increased as compared with the wild-type (0.5 units) 6 h after ConA stimulation (p < 0.05). However, no significant difference was observed after treatment with LPS (Fig. 3).
DNA fragmentation and caspase-3 activity assay
To estimate the resistance against apoptosis, DNA fragmentation and caspase-3 activity assays were performed. At 6 h after cyclohexamide treatment, fragmented DNAs became evident; however, it was difficult to distinguish any visual differences between the bovine Prnp-transfectant and the wild-type. The caspase-3 activity in the transfectant was lower (14.0 ± 0.2 pmol) than in the wild-type (16.3 ± 0.6 pmol) at 24 h after cyclohexamide treatment (p < 0.05) (Fig. 4).
Discussion
These experiments were carried out as basic research for understanding BSE biology through the establishment of an in vitro cellular model. We constituted bovine Prnp-transfectant with the CHO-K1 cell line and examined cellular changes followed by expression of the bovine prion protein. The CHO-K1 cells were transfected with an expression vector containing bovine Prnp ORF, and the transfected cells were selected in a complete medium containing puromycine. The selected cells were then limit-diluted and plated to prepare one cell clone. The primer set used in genomic PCR and RT-PCR was specific for the bovine Prnp introduced into the CHO-K1 cell line, so it could not bind to endogenous hamster Prnp, although the homology of these ORF nucleotides was as high as 82% [21,28].
It was reported that the level of PrPC in the brain represents less than 0.1% of the total amount of protein in the central nervous system; moreover, in other tissues, this concentration is much lower [2]. To detect the expression of bovine prion protein in transfectant, immunoprecipitation was applied using a combination of protein G-coupled agarose and monoclonal antibody 6H4, which has a single linear epitope, DYEDRYYRE, corresponding to positions 144-152 of bovine prion protein [18]. The endogenous hamster prion protein was also detected by immunoprecipitation because hamster PrPC has a similar epitope, differing only in that it has tyrosine at residue 145 rather than the tryptophan of the bovine form [21,28]. The upper band in the Western blot, which was observed only in bovine Prnp-transfectant, seems to be exogenous, while the lower band in CHO-K1 cells might also be endogenous.
PrPC associating with lipid rafts via its GPI anchor has been shown to be a component of the multi-molecular signaling complex, although questions remain as to how the GPI-anchored PrPC, which is present on the extracellular face of the plasma membrane, can directly interact with signaling proteins on the cytosolic face [35].
When assessed by MTT and LDH assays, the bovine Prnp-transfectant showed lower proliferation rates and higher LDH release than the wild-type CHO-K1 cells, which suggests that cell viability is decreased with co-expression of both endo- and exogenous prion protein. Although PrPC in neuronal cells and lymphocytes play an important role in increasing cell proliferation [32,34], CHO-K1 cells transfected with bovine Prnp decreases rather than increases. However, no morphological change was found under microscopic observation.
The LPS-induced production of NO in neuronal cells affected by prion disease has been studied because prions and the major LPS receptor, CD14, are colocalized in lipid rafts through a GPI anchor [20]. Previous research found that, upon treatment with LPS, neuroblastoma N2a cells respond with dose- and time-dependent NO production via increased iNOS mRNA and protein expression. However, in this study, bovine Prnp-transfectant co-expressing endogenous and exogenous prion protein did not show detectable production of NO with LPS or ConA stimulation, and neither did wild-type CHO-K1 cells. This might be due to the use of different types of cells. To induce NO production in CHO-K1 cells, the activation of sphingomyelinase with basic fibroblast growth factor was required to allow the dissociation of the endothelial form of NO synthase from caveolin 1 and its translocation to the cytosol, where it catalyzes the synthesis of NO [10].
Prion protein may have a role in protecting against oxidative stress, and this protection is mediated by Cu/Zn SOD. This has been reported in recombinant, mutant, and normal prion protein in vitro and in vivo [14,23,30,31]. It may be a stress sensor that is sensitive to copper at octapeptide repeats, and it is able to initiate a signal transduction process acting on the antioxidant systems [29]. In addition, the level of the total SOD activity was correlated to the level of prion protein expressed [37]. Our results coincide with other reports that bovine Prnp-transfectant expressing both endo- and exogenous prion protein showed higher SOD activity when stimulated with ConA than do wild-type CHO-K1 cells expressing only endogenous protein. This suggests that the expression of the introduced bovine Prnp aids in the cellular response of the donor CHO-K1 cells to oxidative stress.
It has been reported that prion protein plays a neuroprotective role against apoptosis induced by serum deprivation, and the octapeptide repeat region of prion protein plays an essential role in regulating apoptosis through the activation of SOD and the inactivation of caspase-3/9 [9,16,31]. In our study on apoptosis, caspase-3 activity in bovine Prnp-transfectant lysate was higher than that in the wild-type at 24 h after treatment with cyclohexamide, although it was difficult to establish a visual difference in DNA fragmentation. This indicates that the expression of exogenous bovine prion protein enhanced cellular protection from apoptosis.
In the present study, we described the transfection of the Prnp ORF from Korean cattle into CHO-K1 cells, and the determination of the cellular changes according to different parameters. These cellular changes indicated that the viability of CHO-K1 cells were decreased by the expression of exogenous bovine Prnp, but the cells showed higher cellular protection against antioxidative stress and apoptosis. However, to clarify whether these changes were entirely due to the bovine Prnp or whether there was an additive effect between bovine and hamster Prnp, further experimentation using hamster Prnp-/- and bovine Prnp+/+ neuron cell lines will be necessary. The reconstructed bovine Prnp-transfectant stably expressing the gene might be considered as a profitable tool for further BSE research.



 XML Download
XML Download