

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Colonic motility consists of two major actions, ascending contraction and descending relaxation,and is regulated by the enteric nervous system of the myenteric plexus. Ascending contraction is an intense contraction that propagates aborally through the long segment of the colon. Descending relaxation, allows for rapid propulsion of a large bolus; this is done by giant motor contraction, and prevents the development of tone in the distal segment so that it can accommodate the colonic contents [8]. It has been shown that non-adrenergic, non-cholinergic (NANC) inhibitory neurons, including nitrergic neurons, regulate the descending relaxation phase of peristalsis [13]. NANC relaxation plays an essential role in propulsive motility of the colon [11]. Previous studies have demonstrated that NANC relaxation is significantly blocked by Nω-nitro-L-arginine methyl ester (L-NAME), a non-selective NOS inhibitor, in the proximal and the distal colon [18]. This finding, in the rat, suggests that NANC relaxation is mainly mediated by nitric oxide (NO) in the colon [27], and that endogenous neuronal nitric oxide synthase (nNOS) plays an important role in regulation of intestinal motility [19].
In intestinal inflammatory states, diarrhea is associated with changes in colonic motility and electrolyte transport [21,23]. Clinical studies have suggested that the contractile response is decreased when colon inflammation is present. The lack of contractility is believed to decrease segmental movements of the colonic contents, and accentuates diarrhea in patients with ulcerative colitis [22,26].
Various animal models of experimental colitis have been used to study the effect of inflammation on intestinal motility by employing trinitrobenzene sulfonic acid (TNBS), dextran sulfate sodium (DSS), Trichinella spiralis, or acetic acid as stimulants [9,29]. Since a change of nitrergic neurotransmission was first identified as one of the principal factors causing abnormal motility alteration of nitrergic neurons has been extensively studied. In one model of TNBS-induced colitis, the number of nNOS-immunoreactive nerve fibers was found to be decreased in the circular muscle while nNOS-immunoreactive nerve cell bodies in the myenteric plexus remained unaffected [17]. However, in DSS-induced colitis, both the number of nNOS-immunopositive cells and the activity and synthesis of nNOS were reduced in the myenteric plexus [18]. In a nematode infection colitis model, transient mucosal inflammation decreased NO-mediated relaxation in mice [4]. By contrast, ricin-induced inflammation in the ileum increased NO-mediated inhibition in rabbits [12]. However, alteration of nitrergic neuromuscular transmission has not been studied in an acetic acid-induced colitis model.
Acetic acid causes a mild acute mucosal inflammation in the distal colon of rats [20]. The acetic acid-induced colitis model is an experimental model that has shown morphological similarities to human ulcerative colitis [10]. Acute inflammation is found to be present from 4 h post-acetic acid treatment. The inflammation is characterized by mucosal hemorrhage with a mild mixed inflammatory infiltrate in the lamina propria and submucosal edema. Maximum inflammation is observed from 48 h to 72 h post-acetic acid treatment; it is characterized by a patchy loss of the entire thickness of the crypt epithelium, with a moderate mixed inflammatory infiltrate in the submucosa and edema in the muscularis propria [20]. Therefore, in this study, we used this experimental model of colitis to examine the alteration of nitrergic neuromuscular transmission during inflammation. We studied changes at 4 and 48 h post-acetic acid treatment.
Materials and Methods
Experimental animals
Experiments were performed on male Sprague-Dawley rats (250-300 g). They were housed with free access to food and water and kept under controlled temperature (25℃) and light-dark cycles (12 : 12 h). All experiments were in accordance with the Guide for the Care and Use of Laboratory Animals of Seoul National University.
Induction of colitis
All experimental animals fasted for 24 h before induction of colitis. A distal colitis was caused by intracolonic instillation of acetic acid. This model has been used extensively to investigate the pathogenesis of early phase of inflammation [9,10,31]. Each rat was lightly anesthetized with ether and a polyethylene cannula (PE-60) was inserted into the lumen of the colon via the anus. The tip of the cannula was positioned 8 cm proximal to the anus. Either 1 ml of acetic acid (4% vol/vol in 0.9% NaCl), or saline as the sham control, was slowly infused into the distal colon. After a 30 sec exposure, 1 ml of saline (0.9% NaCl) was injected to completely remove the solution previously infused into the colon. Control animals were studied at 48 h post-saline injection and colitis induced rats were studied at 4 and 48 h post-acetic acid treatment.
Measurement of myeloperoxidase activity
Myeloperoxidase (MPO) activity was measured in the distal colonic tissue obtained from control and rats with colitis. The inflamed distal colon (5 cm) was removed. MPO is an enzyme found primarily in neutrophils; measurement of MPO has been widely used as a marker for intestinal inflammation [25]. MPO activity was measured according to the protocol described by Krawisz et al. [16]. Briefly, after the samples were weighed, tissue samples were homogenized in a buffer (0.5% hexadecyltrimethylammonium bromide in 50 mM potassium phosphate buffer, pH 6.0) for 1 min. The samples were frozen in liquid nitrogen, thawed three times, and centrifuged at 20,000 × g for 20 min at 4℃ using a microcentrifuge. Aliquots of supernatants (20 ml) were mixed with 980 ml of O-dianisidine. Absorbance was recorded at 450 nm every 1 min over a period of 10 min by ELISA. MPO activity was expressed as units/g of tissue. An enzyme unit was defined as the conversion of 1 mol of H2O2 per min at 25℃.
Tissue preparations
Each animal was sacrificed by cervical dislocation; the distal colon was then removed promptly. The colon was opened along the mesenteric border, and the mucosal layer was peeled off and put into a dissecting dish, containing oxygenated (95% O2 and 5% CO2) Krebs solution with the following composition (in mM): NaCl 118, KCl 4.7, KH2PO4 1.2, NaHCO3 25, CaCl2 2.5 and glucose 11. The 2 × 10 mm colonic circular muscle strips were prepared.
Functional studies
One side of the colon strip was pinned to the floor of the recording chamber, and the opposite side was connected to an isometric force transducer (Grass, USA). The strips were allowed to equilibrate for 60 min under an initial tension of 0.1 g. During this period, a bath solution was perfused at a flow rate of 1.5 ml/min, with aerated Krebs solution (95% O2 and 5% CO2). Bath temperature was maintained at 37 ± 0.5℃.
To assess myogenic contractility, each muscle strip was exposed to 10 min of a high concentration (60 mM) of KCl (Sigma, USA) to induce receptor-independent muscle contraction. L-NAME (100 µM; Sigma, USA) was used to investigate the effect of nitric oxide on colonic motility. Electrical field stimulation (EFS: 8 Hz, 120 V, 0.5 ms for 20 sec) was applied, between two-platinium plates, using a Grass S48 stimulator (Warwick; USA) for inducing neuronal response. Tetrodotoxin (TTX, 1 µM; Tocris, UK) was used to confirm that the EFS-evoked responses were neuronal. At the beginning of each EFS experiment, strips were pretreated with atropine (1 µM; Sigma, USA) and guanethidine (5 µM; Sigma, USA). The baseline tension was determined at the end of the experiment by administration of nifedipine (10 µM; RBI, USA).
NADPH-diaphorase histochemistry
Neuronal nicotinamide adenine dinucleotide phosphate (NADPH) diaphorase is a nitric oxide synthase [14]. NADPH-diaphorase histochemistry was performed for detection of nitrergic neurons, which contain neuronal nitric oxide synthase (nNOS). Segments of the distal colon were fixed with 4% paraformaldehyde and 2% picric acid at 25℃ for 4 h. Whole-mounts of longitudinal muscle layers were obtained by peeling off mucosal and circular muscle layers to identity cell bodies of nitrergic neurons in the myenteric plexus [6]. Frozen sections (20 µm thickness) were cut through the segment of the distal colon using a cryostat (Microm, Germany) to identify nitrergic nerve fibers in the circular muscle layer. These two types of specimens were washed in Tris-PBS (pH 7.4), and then reacted in a free-floating state for NADPH-diaphorase histochemistry using β-NADPH (2 mg/ml; Sigma, USA), nitroblue tetrazolium (0.4 mg/ml; Sigma, USA), and 0.3% Triton X-100 in 0.1M with Tris-PBS for 30 minutes at 37℃ [1]. The number of NADPH-diaphorase positive cell bodies was counted under a lightfield microscope (Axioscope; Zeiss, Germany) and the percentage of NADPH-diaphorase positive areas were analyzed using OPTIMAS 5 software (Optimas, USA).
Data analysis
The contractile response to KCl was expressed as mN per cross sectional area (cm2) of the tissue as described previously [30]. Alteration of spontaneous contractions by L-NAME was expressed as the percentage of L-NAME non-treated responses. The time to disappearance of the spontaneous contraction, was calculated as the duration between the initial EFS application, and the reappearance of the first spontaneous contraction during EFS. The number of NADPH-diaphorase positive nerve cell bodies, in the myenteric plexus, was expressed as the number per ganglion, and the NADPH-diaphorase positive area was expressed as a percentage of a selected area. All results are expressed as means ± SE. Statistical analysis was performed by an unpaired Student t test and statistical significance was accepted when p < 0.05.
Results
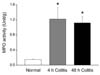
MPO activity
MPO activity has previously been shown to be proportional to the number of neutrophils in the inflamed tissues [16]. Therefore, this was measured from whole distal colonic samples to quantitatively evaluate inflammation at 4 and 48 h post-acetic acid treatment. MPO activity was significantly increased in both the 4 h colitis and 48 h colitis group compared to the normal control colon (Fig. 1).
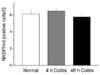
Response to KCl
To investigate whether smooth muscle contractility was altered after induction of colitis, the colonic muscle strips were exposed to 60 mM KCl for 10 min. The response to KCl was not different in colitis groups compared to the normal control group (Fig. 2).
The alteration of phasic contraction by L-NAME
Under NANC conditions, L-NAME (100 µM) was administrated to investigate whether the NO-mediated tonic inhibitory action on the distal colon was altered by acute colitis. In the normal control group, the amplitude of phasic contraction was significantly increased after L-NAME treatment. However, the amplitude of phasic contraction in the colitis groups was not significantly changed after L-NAME treatment. However, the frequency of phasic contraction with L-NAME was not altered after L-NAME treatment in all groups studied (Fig. 3).
Response to EFS
Electrical stimulation was applied under NANC conditions. Spontaneous contractions disappeared during EFS and all responses were completely blocked by the sodium channel blocker, TTX (1 µM). Spontaneous contractions in the normal group were almost completely abolished during EFS (Fig. 4Aa). However, in the colitis group, these contractions initially disappeared, and then reappeared during EFS (Fig. 4Ac and Ae). The time to disappearance of spontaneous contractions during EFS was slightly reduced in the 4 h colitis group and significantly reduced in the 48 h colitis group (Fig. 4B).
To further investigate whether the disappearance of spontaneous contraction during EFS resulted from the nitrergic neurotransmission to the smooth muscle, L-NAME was applied for 10 min before recording the EFS-evoked responses. L-NAME prevented the disappearance of spontaneous contraction during EFS in all experimental groups (Fig. 4Ab, Ad and Af).
NADPH-diaphorase histochemistry
NADPH-diaphorase positive nerve cell bodies were found throughout the myenteric plexus of the colon (Fig. 5). As shown in Fig. 6, the number of NADPH-diaphorase positive nerve cell bodies per ganglion, in the myenteric plexus, was unaltered by acetic acid-induced inflammation of the distal colon. In cross-sections, NADPH-diaphorase positive nerve fibers were identified in the colonic circular muscle layer (Fig. 7). The relative value, of NADPH-diaphorase positive areas in the circular muscle layer, was slightly decreased in the 4 h colitis group and significantly decreased in the 48 h colitis group compared with that of the normal group (Fig. 8).
Discussion
Change of nitrergic neurotransmission is one of the principal causes associated with colitis-induced abnormal intestinal motility. Therefore, the effects of colitis on NOS and nitrergic neuron transmission have been extensively studied in a variety of experimental models. However, the results have differed depending on the animal species studied, the sites of the colon studied, and the manner in which inflammation was induced. The acetic acid-induced colitis model is one of the most useful models to study the pathophysiology of human ulcerative colitis. However, in the acetic acid-induced colitis model, alteration of nitrergic neurotransmission has not yet been reported.
The findings from the present study provide a line of evidence that suggest that nitrergic neuromuscular transmission, to colonic circular muscle, is altered in acetic acid-induced mucosal inflammation. The amplitude of phasic contraction was increased by L-NAME in the presence of atropine and guanethidine in normal tissues. However, the pattern of spontaneous contraction in inflamed tissues was not altered by L-NAME. These results suggest that the amplitude of phasic contraction, in normal colonic tissue, is subject to tonic nitrergic inhibitory control, and that this is impaired in inflamed tissue. Similar observations have been reported in the TNBS-induced rat colitis model where the amplitude of spontaneous contractions increased in conjunction with a loss of nitric oxide synthase activity leading to the reduction of tonic nitrergic inhibition [7].
EFS elicits a response during the stimulation period. Spontaneous contraction disappeared during application of EFS, and this response was completely blocked by TTX; this suggests that the response was mediated via neurons. While the spontaneous contractions almost completely disappeared throughout the EFS application in normal tissues, it initially disappeared and then reappeared during EFS in the acetic acid-induced colitis groups. However, this disappearance was diminished by L-NAME in all groups. Since the disappearance, of spontaneous contractions, was L-NAME-sensitive, it can be interpreted that the EFS-evoked, NO-mediated response was also attenuated in the acetic acid-induced colitis groups. In the DSS-induced rat colitis model, the EFS-induced relaxation in the NANC condition was significantly reduced, suggesting that the decreased relaxation was associated with reduced activity and synthesis of nNOS in the myenteric plexus [18]. In addition, the reduction of nitrergic influence, on the EFS-evoked response, has been reported in TNBS-induced colitis model [32].
Furthermore, it was found that the NADPH-diaphorase reactive area, in the circular muscle layer, was decreased; however, the number of NADPH-diaphorase positive nerve cell bodies in myenteric plexus was not changed in the acetic acid-induced colitis groups. A direct correlation and co-localization between immunohistochemical staining for nNOS immunoreactivity and NADPH-diaphorase histochemical staining have been observed in the nerve plexus of both the small intestine [28] and the large intestine [5]. Therefore, the present result, showing the reduction of NADPH-diaphorase reactivity in the colitis affected circular muscle layer indirectly suggests that nitrergic nerve fibers had degenerated, or that the expression of nNOS in the nerve fibers was suppressed by acetic acid-induced acute inflammation. These findings are consistent with the data obtained from the TNBS-induced colitis model. In this model, a slight decrease in the number of nNOS immunoreactive nerve fibers was observed in the circular muscle layer while the myenteric plexus was unaffected at 2 days post-TNBS treatment [17].
Therefore, the results of the current study show that nitrergic neuromuscular transmission was altered by acute inflammation. It is noteworthy that this alteration was noticeable from 4 hrs post-acetic acid treatment, the reported time at which the inflammatory responses begin to occur in this model [20]. This indicates that nitrergic neurotransmission can be affected by pathological factors involved in the early inflammatory response. In fact, the gene expression of nNOS has been shown to be suppressed by inflammatory cytokines such as interferon gamma and TNF-alpha [2,3]. Therefore, it would be of interest to investigate, in a future study, whether these two cytokines play a major role in the alteration of nitrergic neurotransmission in acetic acid-induced colitis.
The response to KCl did not differ in comparisons between the normal control group and colitis groups. The component related to contraction was unaffected, while the NO-mediated inhibitory response was changed by the presence of inflammation. However, other models [15,20,24] have shown that both nitrergic neuromuscular transmission and muscle contractility were altered. Such differences may be attributed to species used for study as well as methods used to stimulate colitis.
In conclusion, in the current study, circular muscle contraction was not impaired by inflammation. The time to disappearance of spontaneous contraction during EFS, measured as a L-NAME sensitive response, was significantly reduced. This suggests that myogenic contractility was unaffected; however, the NO-mediated neuronal response was impaired by inflammation. Inhibitory nitrergic neural input to the circular muscle was decreased in acetic acid-induced colitis.
This may be attributable to the decrease of the NADPH-diaphorase reactive area in the circular muscle layer. This alteration of nitrergic neuromuscular transmission began to change from the initial stage of inflammation. The current study may contribute to our understanding of the cause of alteration of NO-mediated colonic motility.






 XML Download
XML Download