

 PDF
PDF ePub
ePub Citation
Citation Print
Print


The c-Jun NH (2)-terminal protein kinase (JNK), also called the stress-activated protein kinase (SAPK), is one of three subgroups of the mitogen-activated protein (MAP) kinase family [2]. JNK activation plays essential roles in organogenesis during mouse development by regulating cell proliferation, survival, or apoptosis and in immune responses by regulating cytokine gene expression [10]. The phosphorylation of JNK (p-JNK) may stimulate transcription factors that lead to cell death [14]. JNK activation has been found to be involved in Schwann cell apoptosis induced by serum withdrawal and insulin-like growth factor-I, while the Bcl family proteins rescue Schwann cells, at least in part, by inhibition of JNK activity [3]. The events described above, including oxidative stress, cell proliferation, and cell death/survival, are commonly observed in animals with experimental autoimmune neuritis (EAN), which is an animal model of human peripheral demyelinating diseases such as Guillain-Barré syndrome [4,6].
EAN is a T-cell-mediated autoimmune disease of the peripheral nervous system (PNS) [5]. The clinical course of EAN is characterized by weight loss, ascending progressive paralysis, and spontaneous recovery [1]. It has been proposed that inflammatory mediators produced in the affected sciatic nerve are involved in the pathogenesis of EAN [7,15].
In our previous study, we found that extracellular signal-regulated kinase (ERK) and p38, the member of the MAP kinase family, is activated in EAN lesions in a Lewis rat model [1,8], demonstrating that phosphorylated ERK (p-ERK) [1] and phosphorylated p38 [8] are mainly localized in some Schwann cells as well as in some ED1-positive inflammatory cells in EAN lesions. This observation implies that increased phosphorylation of ERK by autoimmune stimulation is associated with host cell survival, particularly at the late stage of PNS inflammation. Although many studies have focused on the phosphorylation of JNK in Schwann cells in the PNS [3], little is known about the activation of JNK in the pathogenesis of EAN, which is involved in cell apoptosis [11] or survival [10] depending on the stage of cell activation.
The aim of the present study was to elucidate the patterns of JNK phosphorylation in the sciatic nerves of Lewis rats with EAN and to confirm the cell phenotypes that were positive for p-JNK and that may be involved in EAN.
Materials and Methods
Animals
Lewis rats were obtained from Harlan Sparague Dawley (USA) and bred in our animal facility. Thirty-seven female rats aged 7~12 weeks and weighing 160~200 g were used.
EAN induction
Active EAN was induced in the Lewis rats as previously described [1,13]. Twenty-five rats were each injected in both hind footpads with an emulsion containing 100 µg of SP26 (Shimadzu, Japan), which is a peptide homologous to amino acids 53-78 of bovine myelin P2 protein, in complete Freund's adjuvant (CFA) (Mycobacterium tuberculosis H37Ra, 5 mg/ml; Difco, USA) and were evaluated clinically, as previously reported [1]. Each rat was treated with 50 ng of pertussis toxin (Sigma, USA) at days 0 and 2 after immunization. The progress of EAN was divided into four clinical stages: grade 1 (G.1), floppy tail; grade 2 (G.2), ataxia and inability to spread the toes; grade 3 (G.3), paraplegia; and grade 4 (G.4), tetraplegia or moribund condition [7]. At days 10, 14-16, 24, and 30 post-immunization (PI), five rats were killed under ether anesthesia, and 5 cm of the sciatic nerve was removed bilaterally from each. Twelve control rats were immunized with CFA only.
Tissue sampling
To study the phosphorylation of JNK in the sciatic nerves of Lewis rats with EAN, tissues were sampled post-immunization at days 10 (during G.1), 14-16 (during G.3), 24 (after partial recovery of hindlimb paralysis, R.1), and 30 (after recovery of hindlimb paralysis, R.0). The sciatic nerves were obtained from CFA-immunized control rats at each matching day (n = 3 each time point). The sciatic nerves were removed and frozen at -70℃ for protein analysis. Pieces of the sciatic nerve were embedded in paraffin after fixation in 4% paraformaldehyde in phosphate-buffered saline (PBS) at pH 7.4.
Western blot analysis
Each sciatic nerve was homogenized in lysis buffer (40 mM Tris, 120mM NaCl, 0.1% Nonidet p-40, 2mM Na3VO4, 1 mM PMSF, 10 µg/ml aprotinin, 10 µg/ml leupeptin) with 20 strokes in a homogenizer. The homogenates were transferred to microtubes and centrifuged at 12,000 rpm for 20 min, after which the supernatant was harvested. The SAPK/JNK antibody kit (PhosphoPlus; Cell Signaling, USA) was used for the immunoblot assay. The supernatant (containing 40 µg of protein) was loaded onto a 10% SDS-PAGE gel and transferred onto a nitrocellulose membrane (Bio-Rad, USA). The residual binding sites on the membrane were blocked as previously reported [12]. The ratios of p-JNK/total JNK in each group were compared using a one-way ANOVA, followed by a Newman-Keuls post-hoc test. In all cases, differences with p values less than 0.05 were considered significant.
Immunohistochemistry
Paraffin tissue sections (5 µm) of sciatic nerves from control and EAN-affected rats were de-paraffinized and allowed to react with rabbit polyclonal anti-p-JNK (Cell Signaling, USA), rabbit polyclonal anti-S100 (Dako, Denmark), or mouse monoclonal anti-rat macrophage antibodies (ED1; Serotec, UK), with slight modifications from our previous study [9]. The immunoreactions were visualized using avidin-biotin peroxidase complexes (Elite kit; Vector, USA), and the peroxidase reaction was developed using a diamino-benzidine substrate kit (Vector, USA).
For the double staining of two antigens in the same sections, p-JNK and macrophages, double immunofluorescence was applied using tetramethyl rhodamine isothiocyanate (TRITC)-labeled streptavidin (1 : 500 dilution; Sigma, USA) for p-JNK or fluorescein isothiocyanate (FITC)-labeled goat antimouse IgG (1 : 50 dilution; Sigma, USA) secondary antibody for ED1 to co-localize in the same cell.
Double staining of apoptosis and p-JNK, ED1 and S100-immunoreactivity
DNA fragmentation was detected by in situ nick end-labeling, performed according to the manufacturer's instructions (ApopTag; Intergen, USA). The co-localization of the TUNEL (terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling) reaction and p-JNK immunoreactivity was examined by double-labeling in the same section using alkaline phosphatase-labeled avidin (Vector, USA). Co-localization of the TUNEL reaction and either ED1 or S100-immunoreactivity was detected using double immunofluorescent labeling in the same section. In brief, after finishing the TUNEL reaction, which was allowed to react with TRITC-labeled anti-digoxigenin antibody, double immunofluorescence was applied using FITC-labeled goat anti-mouse IgG (1 : 50 dilution; Sigma, USA) or anti-rabbit IgG (1 : 50 dilution; Sigma, USA) secondary antibodies to co-localize the TUNEL reaction and each protein antigen in the same cell.
To reduce or eliminate lipofuscin autofluorescence, the sections were washed in PBS (three times for 1 h) at RT, and then dipped briefly in distilled H2O, and treated with 10 mM CuSO4 in ammonium acetate buffer (50 mM CH3COONH4, pH 5.0) for 20 min, dipped briefly again in distilled H2O, and returned to PBS. The double immunofluorescence-stained specimens were examined with an FV500 laser confocal microscope (Olympus, Japan).
Results
Clinical observation of EAN
The clinical course of EAN was shown in our previous report [1]. In brief, Lewis rats immunized with SP26 peptides developed floppy tails (G.1) at day 10 PI and showed progressive hindlimb paralysis (G.3) at days 14-16 PI. All of the rats subsequently recovered from hindlimb paralysis (R.0) after 24 days PI [1]. Histological examination detected few inflammatory cells at day 14 PI in the sciatic nerve samples from control rats. At days 10-14 PI in the EAN-affected rats, many inflammatory cells were found in the sciatic nerves. The inflammatory cells gradually disappeared in the sciatic nerves at days 24 and 30 PI when animals recovered from hindlimb paralysis [1].
Western blot analysis of p-JNK1/JNK2 in EANaffected sciatic nerves
In the Western blot analysis for p-JNK1 (approximately 46 kDa) and p-JNK2 (approximately 54 kDa) in normal rats, two weak bands were detected in the sciatic nerve. In EAN-affected rats, the level of p-JNK1 was significantly increased at day 14 PI (an increase of 2.39 ± 0.76 [mean ± S.E.] times the normal level, n = 5, p < 0.05), declined after day 24 PI (2.23 ± 0.25, n = 5, p < 0.05), and persisted its expression at day 30 PI (2.11 ± 0.22, n = 5, p < 0.05). With a pattern similar to p-JNK1, the p-JNK2 level was significantly increased at day14 PI (2.37 ± 0.28, n = 5, p < 0.05), showed a significant increase at day 24 PI (3.04 ± 0.25, n = 5, p < 0.01), and declined after day 30 PI (2.31 ± 0.05, n = 5, p < 0.05) in the sciatic nerves of rats with EAN, as compared with levels in normal control rats (n = 5) (Fig. 1).
Localization of p-JNK in EAN-affected rat sciatic nerves
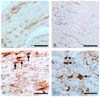
Immunohistochemical analysis was applied to visualize the cell phenotype for p-JNK in the sciatic nerves of normal rats and rats with EAN. In the normal rats, p-JNK was weakly immunostained in some vascular endothelial cells and Schwann cells (Fig. 2A). In the sciatic nerves of rats with EAN lesions, the immunoreactivity of p-JNK was seen in Schwann cells and some inflammatory cells at day 24 PI. There was intense immunostaining for p-JNK in the infiltrating inflammatory cells (Fig. 2C, arrows) and some p-JNK-expressing Schwann cells (Fig. 2D, point-arrows) were also positive.
Double staining of apoptosis and p-JNK, ED1 and S100 expression in the EAN affected sciatic nerves
To match the phosphorylation of JNK to apoptosis of inflammatory cells in EAN-affected sciatic nerves, staining for both p-JNK and the TUNEL reaction was applied. Most of the TUNEL-positive inflammatory cells were immuno-positive for p-JNK in the EAN-affected sciatic nerves at day 24 PI (Fig. 4A, arrows). Some TUNEL reaction was colocalized with ED1 in the sciatic nerves at day 24 PI (Fig. 4B, arrows) as shown in a previous study [4], suggesting that macrophages underwent apoptosis. However, the TUNEL reaction was not detected for the Schwann cells with the present EAN model and immunostaining methods (Fig. 4C).
Discussion
This study is the first to confirm that JNK is phosphorylated in host and inflammatory cells during the course of EAN in a Lewis rat model. Specifically, Western blot analysis revealed that the phosphorylation of JNK was significantly increased in the affected sciatic nerves of animals with EAN.
In a previous study, we found that the expression of p-JNK increased in the spinal cord following encephalomyelitis [12]. As for the role of p-JNK in animals with EAN, we postulate that inflammatory cells, which are ultimately associated with cell death, preferentially express p-JNK [2,11]. For this reason, p-JNK expression declines during the recovery stage of EAN, which is also the stage at which the number of inflammatory cells decline. We also showed in the present study that Schwann cells express p-JNK after stimulation by pro-inflammatory cytokines, as previously reported [3]. In a previous study, Schwann cells were found to undergo apoptosis during EAN [4]. Although we have not found a typical TUNEL reaction on Schwann cells in EAN, we do not exclude the possibility that some Schwann cells were eliminated through apoptosis depending on the EAN model used. We suggest that a majority of Schwann cells during EAN in the present study were rescued from apoptosis through the activation of JNK, as shown in a previous study [3].
In our previous study, we demonstrated that the phosphorylation of ERK increased in the sciatic nerves with EAN at the early stage, and the increased phosphorylation remained in the sciatic nerve even after the EAN rats recovered from paralysis [1]. Therefore, if the results from the earlier study of ERK activation [1] and the phosphorylation of JNK in the present study are taken into consideration, it appears that p-JNK is involved preferentially in the activation of inflammatory cells in the sciatic nerve with EAN, which lead to cell death of inflammatory cells, including ED1-positive macrophages, particularly by apoptosis during the recovery stage of EAN. We postulated that inflammatory cells lose p-JNK signals dynamically and gain other signals, such as iNOS [7], to lead to apoptosis during EAN.
When all of the information is considered, we confirmed in the present study that the expression of the phosphorylated form of JNK is transiently increased in the sciatic nerve following the onset of EAN. This suggests that temporal increase of p-JNK in inflammatory cells and Schwann cells in animals with EAN may differentially influence cell signaling depending on cell types, which is crucial for the modulation of inflammation during the course of autoimmune PNS diseases, including human Guillain-Barré syndrome and its animal model EAN.



 XML Download
XML Download