

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Pasteurella multocida is a Gram-negative bacterium that has been identified as the causative agent of several economically important diseases affecting different species of animals [30]. The bacteria can be classified into 5 serotypes (A, B, D, E, and F) and 16 serogroups (1–16) based on capsule structure and lipopolysaccharide (LPS) component, respectively [32]. Hemorrhagic septicemia (HS) is an acute and fatal septicemic disease of large ruminants caused by P. multocida serotype B:2. The disease is mainly distributed in Asia and Africa and affects cattle and buffalo [49]. HS mainly occurs during typical monsoon periods with high humidity and temperature in Asia [9]. Stressful conditions, such as those associated with climate change, transportation, and housing management, increase an animal's glucocorticoid levels. Glucocorticoid, a stress-related hormone, may suppress the immune defense mechanisms, which can increase the susceptibility of animals to diseases [21428]. Dexamethasone (DEXA), a synthetic glucocorticoid, has been experimentally used to suppress the immune system of the animals [3]. In contrast, LPS is an endotoxin originating from the outer membrane of Gram-negative bacteria. LPS is an immunostimulatory agent that stimulates the immune response of animals. A previous study has shown that LPS of P. multocida serotype B strain is able to stimulate humoral immunity in mice [29]. Furthermore, buffalo inoculated with purified LPS of P. multocida B:2 developed clinical signs of HS [16]. Meanwhile, LPS of P. multocida serotype A is an essential virulence factor in fowl cholera. Harper et al. [15] demonstrated that mutants of P. multocida serotype A:1 strain VP161, which lacked the outer core structure and LPS, were completely attenuated and unable to infect a chicken host.
The tonsillar region, an area that allows P. multocida B:2 to acquire nutrients and replicate, is believed to be an initial site of HS infection. For the infection to succeed, the bacteria may release toxins or interact with the host cells [591020]. Upon successful infection, the bacteria are able to translocate from the mucosal cells of the tonsil and eventually enter the bloodstream, which results in a rapid spread of septicemia in the animal. Several previous studies have found that the P. multocida A:3 strain, A:3,4 strain, B strain, and B:2 strain invade chicken embryonic fibroblast cells [1], primary turkey kidney cells [21], bovine aortic endothelial cells (BAECs) [12], and embryonic bovine lung cells [25], respectively. In addition, Galdiero et al. [12] and Othman et al. [25] have suggested that P. multocida serotypes B and B:2 JRMT12 invade bovine cells by utilizing a microfilament-formation-dependent invasion mechanism. However, factors that influence that invasion mechanism are still not fully described. This study aims to investigate the invasion efficiency of P. multocida serotype B:2 on BAECs that are treated with DEXA or LPS, and to characterize the effect of those treatments on cellular actin filaments.
Materials and Methods
Bacterial strain
The P. multocida serotype B:2 was kindly provided by associate professor Dr. Faez Firdaus Jesse Abdullah, Faculty of Veterinary Medicine, Universiti Putra Malaysia. The bacteria were isolated from an outbreak of HS in Kelantan, Malaysia [18]. The bacteria were subcultured on blood agar with 5% sheep blood (Oxoid, UK) and incubated at 37℃. To propagate the bacteria, a single colony from the blood agar was inoculated in brain heart infusion (BHI) broth (Pronadisa, Spain) and cultured at 37℃ with shaking at 50 × g in an SI-600R incubated shaker (Jeio Tech, Korea).
Cell culture
BAECs (BW-6002, lot no. 8F3173; Lonza, USA) were grown in a growth medium containing endothelial cell basal medium (EBM CC-3121; Lonza) that was supplemented with 10% fetal bovine serum (FBS) (American Type Culture Collection, USA), 0.4% (v/v) bovine brain extract, 0.125% (v/v) hydrocortisone, 0.125% (v/v) gentamicin and sulphate amphotericin-B, and 0.1% (v/v) human recombinant epidermal growth factor in a buffered bovine serum albumin saline solution (EGM-MV SingleQuots CC-4143; Lonza). The BAECs were incubated in a humidified atmosphere of 5% (v/v) CO2 in an incubator (Binder, Germany) at 37℃.
Cell viability assay
BAECs were counted via the trypan blue exclusion method as described in a previous study [25]. The cells (~1 × 105) were seeded in 96-well tissue culture plate (TPP, Switzerland) and incubated overnight at 37℃ in the 5% (v/v) CO2 incubator. The growth medium without FBS was used as a diluent for DEXA (Calbiochem, Germany) and LPS (Sigma, USA). After washing twice with phosphate-buffered saline (PBS; Life Technologies, USA), the cells in each well were treated with 100 µL of 0.1, 1, 10, 100, or 1,000 µM of DEXA or 25, 50, 100, 200, 500, or 1,000 ng/mL of LPS. The cells were incubated in the 5% (v/v) CO2 incubator at 37℃ for 24 h. To each well was added 10 µL of 5 mg/mL of MTT solution (Life Technologies) and the plate incubated in the 5% (v/v) CO2 incubator at 37℃ for 3 h. Subsequently, the media were removed, and 100 µL of DMSO (Fisher Scientific, UK) were added to each well. The plate was incubated in the 5% CO2 (v/v) incubator at 37℃ for 30 min. The absorbance (optical density [OD]) of each well was determined at 540 nm by using a microplate reader (Bio-Tek, USA). Three independent assays with six replicate samples each were performed. The viability of the cells was calculated as viability (%) = (ODtreated/ODuntreated) × 100.
Invasion assay
Invasion assays were carried out according to a method described by Othman et al. [25] with modification. Briefly, the bacteria were harvested from 18 h cultures in BHI broth by centrifugation at 2,000 × g for 5 min and then resuspended in PBS. The BAECs were seeded onto a 6-well tissue culture plate (TPP) and incubated in the 5% (v/v) CO2 incubator at 37℃ overnight. After a washing step with PBS, the cells in each well were treated with 1, 10, or 100 µM of DEXA or 25, 50, or 100 ng/mL of LPS and incubated in the 5% (v/v) CO2 incubator at 37℃ for 24 h. The viability of the cells was examined by applying the trypan blue exclusion method. The bacterial suspension was adjusted with the growth medium without FBS to a multiplicity of infection of 100:1 (bacteria:mammalian cell) based on an OD that represents 2 × 109 colony-forming units (CFU)/mL. After a washing step with PBS, the bacterial suspension was added to each well. The plate was centrifuged at 1,000 × g for 5 min (Centrifuge 5810 R; Eppendorf, Germany) and incubated in the 5% (v/v) CO2 incubator at 37℃ for 2 h. After washing three times with PBS, a mixture of 50 µg/mL polymyxin B sulphate (Calbiochem) and 50 µg/mL gentamicin (Amresco, USA) in growth medium without FBS was added to each well of the plate and the mixture incubated in the 5% (v/v) CO2 incubator at 37℃ for 1 h to kill the extracellular bacteria. The cells were then trypsinized by the addition of 1 mL of 0.25% (w/v) trypsin-EDTA (Sigma) at 37℃ for 5 min. The cell suspension was harvested into 15 mL centrifuge tube by centrifugation at 150 × g for 5 min at room temperature. The pellet was resuspended in 900 µL of growth medium without FBS, and the viable cells were counted by using the trypan blue exclusion method. The remaining cells were lysed by the addition of 100 µL of digitonin (Calbiochem) to 100 µg/mL and incubated in the 5% (v/v) CO2 incubator at 37℃ for 30 min. The intracellular bacteria were counted by the viable count plate method. Three independent assays with triplicate samples in each assay were conducted.
Quantification of bacteria by real-time PCR
The real-time polymerase chain reaction (qPCR) assay was performed to detect extracellular and intracellular bacteria subsequent to performing the invasion assay as described above. The genomic DNA of the bacterial culture was extracted by using the DNeasy Blood & Tissue Kit (Qiagen, Germany) according to manufacturer's instruction. The primers used in this study, PmHS forward (5′-CCTTCTACACAAGSTGGTTTGA-3′) and PmHST Reverse (5′-GCCAARTCATTACCRTCA-3′) amplified 178 base pairs (bp) of the est gene of HS-causing P. multocida as described recently by Petersen et al. [26]. The est gene of P. multocida B:2 strain PMTB has been recently identified and characterized in silico, under taxid 1220026 [33]. Ten-fold serial dilutions of the bacterial DNA were made from 2.7 pg to 270 ng to construct a standard curve. Milli-Q water (EMD Millipore, Germany) was used as a non-template control, and the DNA of uninfected BAECs was used as a negative control.
The qPCR was run with 1 × iQ SYBR Green Supermix (Bio-Rad, USA), 1.25 µM of both primers, the bacterial DNA template, and Milli-Q water to create a final volume of 20 µL. The running conditions were an initial denaturation at 95℃ for 3 min, followed by 40 cycles of denaturation at 95℃ for 20 sec, annealing at 54℃ for 45 sec. and extension at 72℃ for 30 sec. The running conditions were performed by using a CFX96 qPCR detection system (Bio-Rad). Data were analyzed by using Bio-Rad CFX Manager software (ver. 3.1; Bio-Rad). Each assay was carried out independently two times with triplicate samples in each assay.
Transmission electron microscopy
Treated and untreated BAECs, as described in the invasion assay, were examined via transmission electron microscopy (TEM). Briefly, the cell mixtures were fixed in 2.5% (v/v) glutaraldehyde (Agar Scientific, UK) in 0.1 M cacodylate buffer for 6 h at 4℃. The cell suspension was centrifuged for 5 min at 1,677 × g using a Minispin (Eppendorf), and the pellet was incubated with 1 mL of horse serum (Faculty of Veterinary Medicine, Universiti Putra Malaysia) overnight. The cubic clotted samples were fixed in 2.5% (v/v) glutaraldehyde for 2 h at 4℃ and washed three times with 0.1 M sodium cacodylate buffer (Agar Scientific) before fixing in 1% (w/v) osmium tetroxide (Agar Scientific) for 2 h at 4℃. The fixed samples were washed again with 0.1 M sodium cacodylate buffer. The samples were then dehydrated with graded acetone 35% to 100% and infiltrated with graded epoxy resin 50% to 100%. Finally, the samples were embedded in beam capsules with resin. Ultrathin sections were obtained with an ultramicrotome and the sections were stained with 2% (w/v) uranyl acetate (Agar Scientific) and lead citrate (Agar Scientific). The samples were observed and examined under a TEM (H-7100; Hitachi, Japan) operating at 100 kV.
Differential immunostaining of intracellular and extracellular P. multocida B:2
BAECs were seeded onto sterile coverslips in a 6-well tissue culture plate in a final volume of 2 mL growth medium with 10% FBS. After washing three times with PBS, the cells were treated with 10 µM of DEXA or 100 ng/mL of LPS in each well with a final volume of 2 mL in growth medium without FBS and incubated in the 5% (v/v) CO2 incubator at 37℃ for 24 h. PBS containing 1% (w/v) bovine serum albumin (Sigma) was used as a blocking buffer and as a diluent for rabbit serum and immunofluorescent dyes. All solutions and incubation in the experiment were conducted on ice. The cells were fixed with 3.7% (v/v) formaldehyde (R&M Chemicals, UK) for 15 min and then incubated with a blocking buffer for 15 min. Polyclonal rabbit serum containing anti-P. multocida B:2 as a primary antibody was kindly provided by Dr. Salleh Annas, Faculty of Veterinary Medicine, Universiti Putra Malaysia. The cells were incubated with the primary antibody (1:200) for 1 h. After washing three times with PBS, the cells were incubated with Alexa Fluor 488 conjugated polyclonal goat anti-rabbit IgG antibody (1:500; Life Technologies) in the dark for 1 h to stain the extracellular bacteria. After washing with PBS, the cells were permeabilized via the addition of 0.1% (v/v) Triton X-100 (Life Technologies) in PBS for 15 min and then incubated with a blocking buffer for 15 min. The cells were then incubated with primary antibody for 1 h. After washing three times with PBS, the cells were incubated with Alexa Fluor 633 conjugated polyclonal goat anti-rabbit IgG antibody (1:500; Life Technologies) in the dark for 1 h to stain both the intracellular and extracellular bacteria. Lastly, the cells were then washed three times with PBS and once with Milli-Q water. The slides were mounted with ProLong Gold antifade with DAPI (4',6'-diamidino-2-phenylindole; Life Technologies) and sealed with nail polish to prevent dehydration. The slides were examined by using an Olympus IX81 microscope (Olympus, USA).
Quantification and fluorescent staining of F- and G-actin
BAECs were seeded onto sterile coverslips in a 6-well tissue culture plate. The cells were treated with 10 µM DEXA or 100 ng/mL LPS and prepared for staining as described in immunostaining assay. The cells were fixed with 3.7% (v/v) formaldehyde (R&M Chemicals) in PBS for 10 min. After washing three times with PBS, the cells were permeabilized by addition of 0.1% Triton X-100 (Fisher Scientific) in PBS for 5 min. After a washing step as described in immunostaining assay, the cells were incubated with 1 unit/mL of Alexa Fluor 488 conjugate phalloidin (Life Technologies) to stain the F-actin or with 9 µg/mL of Alexa Fluor 594 conjugated deoxyribonuclease I (Life Technologies) to stain the G-actin in the dark for 20 min. The cells were washed with PBS followed with a final wash with Milli-Q water. The slides were mounted with ProLong Gold antifade with DAPI and sealed with nail polish to prevent dehydration. The slides were examined by using the Olympus IX81 microscope. The fluorescent units per area of the F- and G-actin were evaluated by using ImageJ software (ver. 1.46r; National Institutes of Health, USA). Each assay was performed independently two times with at least ten replicates in each assay.
Results
Effect of DEXA and LPS on viability of BAECs
The cells showed 87% viability when treated with DEXA at 10 µM for 24 h and showed a dose-dependent decreasing trend in cell viability (panel A in Fig. 1). Meanwhile, the cells showed 69% viability when treated with LPS at 25 ng/mL for 24 h (panel B in Fig. 1). Overall, the cells treated with a DEXA concentration lower than 100 µM showed a cell viability of more than 70%, while cells treated with an LPS concentration lower than 100 ng/mL showed a cell viability of more than 65%.
Invasion of P. multocida B:2 in BAECs
We found that P. multocida B:2 was invasive in both treated and untreated BAECs. Moreover, the invasion efficiency of the bacteria in DEXA-treated cells was not significantly different (p > 0.05) than that of untreated cells (Table 1). The results also showed that P. multocida B:2 invasion efficiency was significantly higher (p < 0.001) in LPS-treated cells at 50 and 100 ng/mL than that in untreated cells (Table 1).
Quantification of P. multocida B:2 in BAECs by qPCR
A standard curve was constructed by using 10-fold serial dilutions of genomic DNA of P. multocida B:2 (panel A in Fig. 2). The qPCR assay was able to detect 102 to 107 CFU of bacteria with quantification cycle values in the range of 12 to 29, and thus, it was used to enumerate the intracellular bacteria in the invasion assay. The primers failed to amplify the DNA from BAECs (panel B in Fig. 2). The cells treated with LPS had a significantly higher number of bacteria per cell (0.01 < p < 0.001) than the DEXA-treated and untreated cells. However, the increase in the number of bacteria per cell of DEXA-treated cells compared to untreated cells was not significant (panel B in Fig. 2).
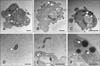
Visualization of intracellular P. multocida B:2 by TEM
Panels A, B, and E in Fig. 3 show the presence of intracellular bacteria in the DEXA-treated and untreated cells, with the bacteria residing in the vacuolar compartment of the cells. However, the intracellular bacteria were observed to reside within the plasma membrane of the LPS-treated cells as shown in panels C and F in Fig. 3.
Visualization of intracellular and extracellular P. multocida B:2 by fluorescence microscopy
The merged images (Fig. 4) reveal the presence of intracellular bacteria in both treated and untreated cells. We observed that the number of intracellular bacteria in LPS-treated cells was higher than that in DEXA-treated and untreated cells. The results indicate that the bacteria are able to invade both treated and untreated cells.
Effect of DEXA and LPS on F- and G-actin pools of BAECs
In Fig. 5, the graph reveals that LPS increased the G-actin pool of the cells (p < 0.001), whereas it decreased the F-actin pool of the cells (p < 0.001). However, the F- and G-actin pools of the DEXA-treated cells were not significantly different from those of the untreated cells (p > 0.05). In addition, the DEXA- and LPS-treated cells had similar total F- and G-actin pools compared to those of the untreated cells. These results indicate that LPS can disorganize the actin filaments of BAECs through a decrease in the F-actin pool and an increase in the G-actin pool, whereas DEXA failed to exert significant changes on the actin filaments of BAECs compared with untreated cells.
Visualization of DEXA- and LPS-affected actin filaments of BAECs
LPS-treated cells showed less density of the filamentous network of F-actin than that of DEXA-treated and untreated cells, whereas the LPS-treated cells showed a higher density of G-actin than that in DEXA-treated and untreated cells (Fig. 6). These results suggest that LPS destabilizes the actin filaments of BAECs by decreasing the F-actin pool and increasing the G-actin pool.
Discussion
Several studies have shown that the endotoxin and LPS of P. multocida B:2 have important roles in the pathogenesis of HS in buffalo and cattle, in which inoculation of LPS is able to reproduce typical symptoms of HS including an increase in the serum level of tumor necrosis factor-alpha (TNF-α) and infiltration of inflammatory cells in various organs, namely lungs, lymph nodes, spleen, gastrointestinal tract, liver, kidney, and heart [816]. Furthermore, P. multocida LPS is able to induce the release of proinflammatory and immunomodulatory cytokines such as interleukin 1 alpha (IL-1α), IL-6, TNF-α, interferon alpha, and IL-12 from murine splenocytes [17]. However, the association between LPS and the in vitro invasion ability of P. multocida has not been reported. In this study, we observed that BAECs treated with LPS at 100 ng/mL for 24 h had a higher invasion efficiency (p < 0.001) of intracellular P. multocida serotype B:2 compared with DEXA-treated and untreated cells. In addition, DEXA-treated cells showed an invasion efficiency similar to that of untreated cells. This result indicates the importance of LPS in P. multocida serotype B:2 invasion of cells, and, eventually, its entry into the bloodstream resulting in septicemia. Further study needs to be carried out to confirm this hypothesis.
Our results also suggest that treatment with LPS but not with DEXA can induce depolymerization of actin filaments of BAECs. This observation agrees with Chakravortty et al. [6] who reported that LPS reduced the F-actin pool and increased the G-actin pool, which indicates F-actin depolymerization and disorganization of the actin filaments of the cells. Furthermore, studies have shown that P. multocida serotype B:2 invade embryonic bovine lung cells via the microfilament-dependent mechanism [1225]. Hence, one possible explanation for the results of the present study is that LPS affects the actin filaments of BAECs, which allows a higher number of P. multocida B:2 to enter the cells. The actin filament changes would allow the bacteria to interact with the cells and facilitate bacterial entry into host cells. However, BAECs treated with 1 µg/mL cytochalasin D, an inhibitor of actin polymerization, for 24 h showed poor fluorescence images due to detachment of the cells (data not shown). Hence, based on our results we are only able to speculate that the microfilament changes after treatment could affect the invasive capability of P. multocida B:2 into BAECs. Further study is required to establish the role of actin microfilaments during the invasion of P. multocida into cells.
West et al. [31] have reported that the LPS and the type III secretion system of Shigella have roles in mediating bacterial invasion into host cells by inducing cell membrane damage and reorganizing the actin cytoskeleton. A study carried out by Chakravortty et al. [6] indicated that LPS induced injury of BAECs directly and in the absence of non-endothelial cell mediators in an in vitro culture system. Hence, damage or alteration of BAECs could be possible when the cells are treated directly with LPS. In the absence of immune cells, LPS could damage or alter the mechanisms of the cells that facilitate bacterial uptake. In this study, we only performed the invasion assay with P. multocida; thus, it is unclear whether a similar effect can be demonstrated with other bacteria, including non-pathogenic bacteria. Since LPS is able to induce actin depolymerization and increase vascular permeability to various macromolecules [13], it is very likely that LPS might promote the cellular entry of non-pathogenic bacteria.
The treatment of BAECs with DEXA and LPS may influence the viability of the cells in a dose-dependent manner. A previous study by Chen et al. [7] has shown that DEXA at concentrations of 100 µM and 1,000 µM decreased cell proliferation and increased cell apoptosis in bovine corneal endothelial cells. In this study, BAECs treated with 1 and 10 µM DEXA showed > 70% cell viability (panel A in Fig. 1) and should able to support bacteria entry, if any, whereas the LPS-treated cells at 100 ng/mL for 24 h showed > 65% of cell viability (panel B in Fig. 1). These results are similar to those in a report by Frey and Finlay [11], which showed the percentage of cell death of bovine pulmonary artery endothelial cells was approximately 40% following LPS treatment at 100 ng/mL for 24 h.
In this study, the number of intracellular bacteria following the invasion assay was measured based on conventional and qPCR assays, and both assays showed an increase in the intracellular bacteria following treatment of BAECs with LPS (0.01 < p < 0.001). In the report by Nadkarni et al. [24] using the viable plate count method and real-time PCR assay to enumerate the bacterial number of Escherichia coli, Pseudomonas aeruginosa, and Staphylococcus aureus, both methods yielded similar estimates of bacterial number. Furthermore, Macé et al. [22] have reported that a real-time PCR assay had a higher accuracy and reliability than that of a viable plate count method in the quantification of Photobacterium phosphoreum bacteria in salmon. Hence, the results of the qPCR assay in this study were comparable with the results of the viable plate count method.
Based on the TEM results, the bacteria were observed in the vacuoles of the DEXA-treated and untreated cells, whereas the bacteria internalized within the plasma membrane of the LPS-treated cells. Similar observations in which P. multocida serotype B were observed within the phagocytic vacuoles of untreated BAECs and embryonic bovine lung cells have been reported [1225]. In this study, the intracellular bacteria were internalized within the plasma membrane of the LPS-treated cells in contrast to DEXA-treated and untreated cells, which were found primarily in the vacuoles. That difference is probably due to the LPS treatment destabilizing the actin filament of the cell membrane of BAECs, which could facilitate bacterial invasion. Another possible mechanism that facilitates such invasion is the activation of the P. multocida B:2 gene(s) that are involved in the actin-dependent invasion of the bacteria into host cells.
A differential fluorescence-labeling method based on Mooney et al.'s report [23] on visualizing intracellular bacteria within host cells was adopted in this study. DEXA-treated cells showed a similar ratio of intracellular bacteria per cell as that of untreated cells, whereas LPS-treated cells had a higher number of intracellular bacteria per cell compared to the DEXA-treated cells. Those results are in close agreement with the those of invasion assay in this study indicating that LPS-treated cells might enhance the invasive capability of P. multocida B:2 to its host cell. Kyoui et al. [19] reported that the internalin A gene (inlA) of Listeria monocytogenes has an important role in cell invasion by regulating attachment to host cells. Meanwhile, the virulence gene ActA of L. monocytogenes is essential in bacterial actin-based motility by regulating catalysis in the assembly of filamentous actin network for actin tail formation after infection of the host cells [27]. Further studies are required to identify the P. multocida B:2 gene(s) that are involved in the actin-dependent invasion of bacteria into the host cells. Nevertheless, it seems that actin polymerization is a part of a mechanism related to bacterial entry into cells or bacterial uptake by cells.
In conclusion, our findings suggest that LPS has an important role in increasing the ability of P. multocida B:2 to invade cells by modulating the actin microfilaments of the infected cells, a key step in bacteria gaining access to the bloodstream and, subsequently, causing septicemia. However, further studies based on in vivo infection of susceptible animals should be carried out to confirm the actual mechanisms of LPS involved in modulating HS infection and progression.






 XML Download
XML Download