

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Since the detection of highly pathogenic avian influenza (HPAI), A/goose/Guangdong/1/1996 (Gs/Gd), in Southern China, Gs/Gd lineages are now widely distributed in poultry and wild birds in Asia, Europe, the Middle East, and Africa [14]. In South Korea, there have been five major HPAI epidemics involving the H5 HPAI viruses between 2003 and 2016 [78121315].
In January 2014, novel H5N8 viruses were identified for the first time to be the cause of an HPAI outbreak in South Korea. Those viruses were related to the H5N8 virus circulating in domestic poultry in Eastern China [13]. The viruses belonged to the H5 clade 2.3.4.4 and were classified into two genetic groups, group A and group B, with group A predominant in South Korea [413]. Group A viruses were further classified into four subgroups: C1, C2, C3, and C4. The H5N8 viruses circulating in domestic poultry in South Korea since September 2014 are from the C1 subgroup. Viruses in the C2, C3, and C4 subgroups have spread globally after circulating among wild birds in breeding sites such as those in Siberia and Beringia [10]. The C2 and C4 strains resulted in HPAI outbreaks in Asia and Europe/Asia, respectively, whereas C3 strains have been primarily detected in North America, Japan, and Taiwan [210].
To identify epidemiological features in South Korea and the genetic evolution of H5N8 viruses during the HPAI outbreak period, the geographical and chronological distributions of H5N8 viruses were investigated by undertaking phylogeographical analyses, and the results were compared with the distribution of H5N8 viruses from other countries.
Samples from feces, oropharyngeal and cloacal swabs, and carcasses were collected from wild birds and poultry via active and passive surveillance during the 2015 to 2016 H5N8 HPAI outbreak. Viruses were isolated by inoculating samples into 10-day-old specific pathogen free (SPF) embryonated chicken eggs. The allantoic fluids from the inoculated SPF eggs were screened by using the hemagglutination (HA) assay and RT-PCR. Of the 153 viruses isolated from January 2015 to April 2016, five isolates from wild birds and 39 isolates from poultry were selected, based on sampling locations and collection dates, for genetic evolution analysis (Table 1).
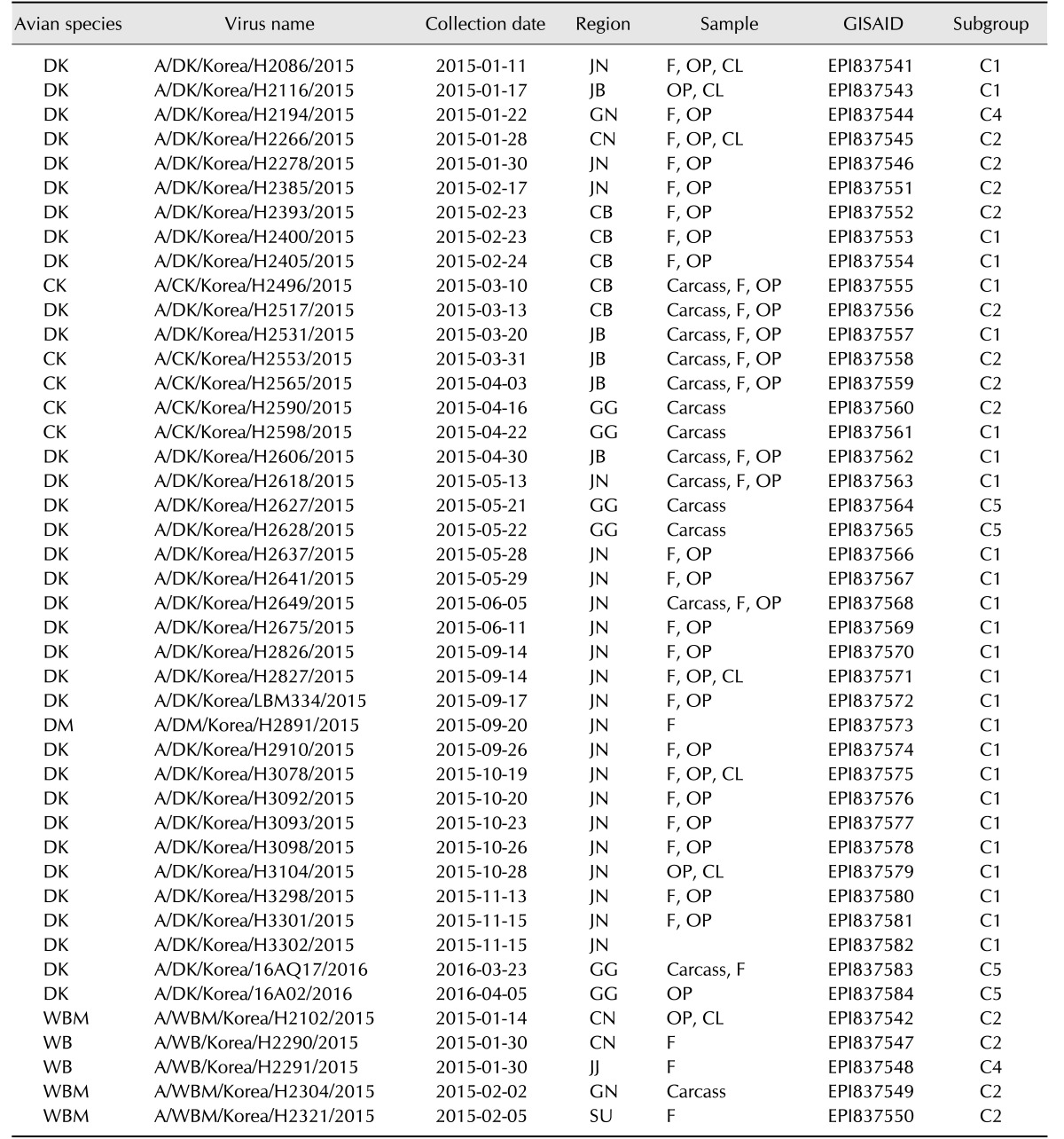
Viral RNA was extracted from allantoic fluid by using the Viral Gene-spin viral DNA/RNA extraction kit (iNtRON Biotechnology, Korea). The HA gene of H5N8 viruses was amplified with HA gene-specific universal primers [3] by using the One-step RT-PCR kit (Qiagen, USA). The PCR products were purified by using the QIAquick Gel extraction kit (Qiagen) and directly sequenced (Cosmogenetech, Korea) with an ABI 3730 genetic analyzer (Applied Biosystems, USA). The global initiative on sharing all influenza data (GISAID) accession numbers of the new viruses isolated between January 2015 and April 2016 are EPI837541–EPI837584.
HA sequences from 57 H5N8 isolates from South Korea in 2014 and 24 H5 sequences from various countries during 2014 and 2015 were obtained from the Influenza Research Database (National Center for Biotechnology Information, USA) and GISAID (GISAID Platform, Germany). The CLC Main Workbench software (ver. 6.8.2; CLC bio, USA) was used to edit and align sequences. For analysis of the geographic spread of H5N8 viruses and its phylogenetic tree, BEAST v1.8.1 (BEAST, Drummond AJ, Suchard MA, Xie D and Rambaut A) was used in combination with BEAGLE v2.1 (BEAGLE Library; GitHub, USA). Analyses were performed under the SRD06 nucleotide substitution and Bayesian skyline coalescent models. Discrete phylogeographical analyses were performed by using the Bayesian stochastic search variable selection. Four Markov chain Monte Carlo simulations were independently run for 100 million steps, with sampling every 10,000 steps. Outputs and effective sample size were examined by using Tracer v1.6 (BEAST, Rambaut A, Suchard MA, Xie D and Drummond AJ). Outputs were combined with 10% burn-in, and a maximum clade credibility (MCC) tree was generated by using TreeAnnotator v1.8.1 (GitHub) in BEAST. Phylogeographical trees were visualized by using FigTree v1.4.2 (Molecular Evolution, Phylogenetics and Epidemiology, Rambaut A).
According to the timeline of virus detection, there were four H5N8 outbreaks between January 2014 and April 2016. In that period, 393 H5N8 viruses in poultry and 58 in wild birds in South Korea were isolated via active and passive surveillance. The majority of those viruses were detected in western part of Korea, which has abundant poultry farms and wintering habitats for migratory birds such as the Baikal teal, mallard, bean goose, and spot-billed duck (panel A in Fig. 1) [2]. Furthermore, H5N8 viruses were detected in poultry, as well as in wild birds, in early and late 2014 (panel B in Fig. 1). According to a previous study [4], the H5N8 outbreak on poultry farms in early 2014 was of geographical relevance as the poultry sites were near where H5N8 was detected in infected wild birds. Therefore, by undertaking active surveillance in wild birds, early detection of HPAI virus can be helpful in decreasing the likelihood of virus introduction into poultry farms.
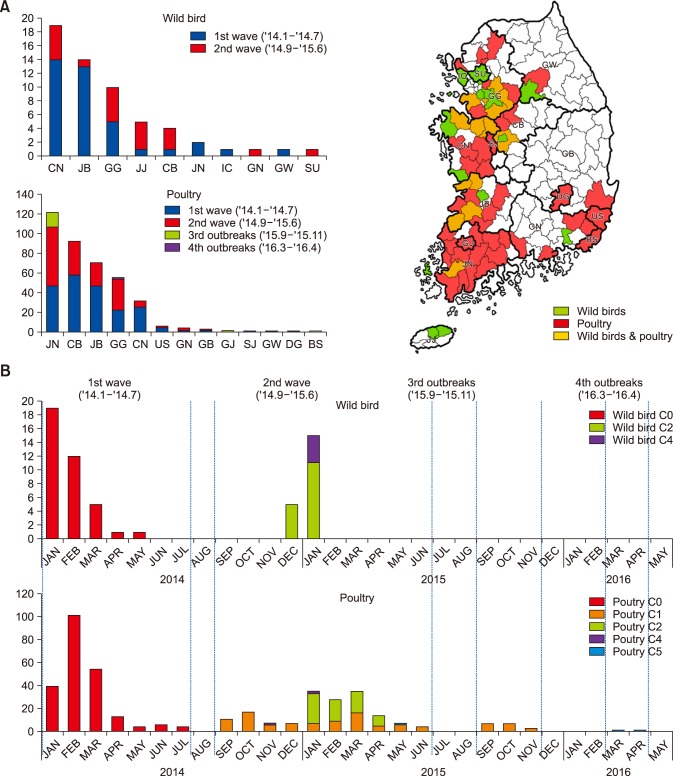 | Fig. 1Geographical and chronological distribution of H5N8 viruses in wild birds and poultry. (A) Bars represent the number of H5N8 virus isolates in wild birds (upper) and poultry (lower) according to the provinces of South Korea between January 2014 and April 2016. The map of South Korea indicates the provinces and species in which H5N8 viruses were isolated: wild birds (green), poultry (red), and wild birds and poultry (orange). JB, Jeonbuk; JN, Jeonnam; CB, Chungbuk; CN, Chungnam; GG, Gyeonggi; GN, Gyeongnam; JJ, Jeju; SU, Seoul; SJ, Sejong; GJ, Gwangju; IC, Incheon; GW, Gangwon; US, Ulsan; DG, Daegu; and GB, Gyeongbuk. (B) Bars represent the number of H5N8 virus isolates in wild birds (upper) and poultry (lower) according to time and virus subgroups from January 2014 to April 2016. Red, C0 subgroup; orange, C1 subgroup; green, C2 subgroup; purple, C4 subgroup; and blue, C5 subgroup.
|
The majority of H5N8 viruses (> 95%) from poultry were isolated in the Jeonnam (JN), Chungbuk (CB), Jeonbuk (JB), Gyeonggi (GG), and Chungnam (CN) provinces, which are located in western part of Korea (panel A in Fig. 1). At least 73% of the H5N8 viruses were isolated in domestic ducks and, of those, approximately 66% were from the JN, CB, and JB provinces, which comprises more than 84% of the domestic duck population in South Korea (data not shown). Ducks infected with H5N8 virus shed abundant amounts of virus via the respiratory and intestinal tract without severe clinical signs [5]. This suggests that the high density of duck farms with low biosafety levels was a major factor in the largest outbreak of H5N8 in South Korea.
Over the 2014–2016 H5N8 outbreak period, the viruses have further evolved into multiple subgroups that have antigenic characteristics including the same HA cleavage site and the typical avian configuration; E186, G221, Q222, G224 based on H5 numbering. The primitive H5N8 viruses (C0 group) have continuously circulated in major and minor domestic poultry farms in Korea and have evolved into the C1 and C5 groups (panel B in Fig. 1 and Fig. 2) via an accumulation of point mutations. The C1 group viruses were frequently detected on major poultry farms in the JN province from September 2014 to November 2015 (panel B in Fig. 1 and Fig. 2). Viruses in the C5 subgroup have been detected sporadically and, for the first time, were detected on duck farms in the GG province in May 2015. Two further cases were detected in March and April 2016 on duck and minor poultry farms in the same province during intensive active surveillance (panel B in Fig. 1 and Fig. 2). The results of the phylogeographical analyses reveal long branches in the C5 group (Fig. 2). Those results suggest that H5N8 viruses in the C5 subgroup have consistently circulated in poultry farms, most likely in minor poultry farms, but were undetected for a long time. The route of transmission among poultry farms remains unknown. Since May 2016, H5N8 has not been detected through intensive surveillance, and South Korea declared itself to be free from HPAI as of August 2016.
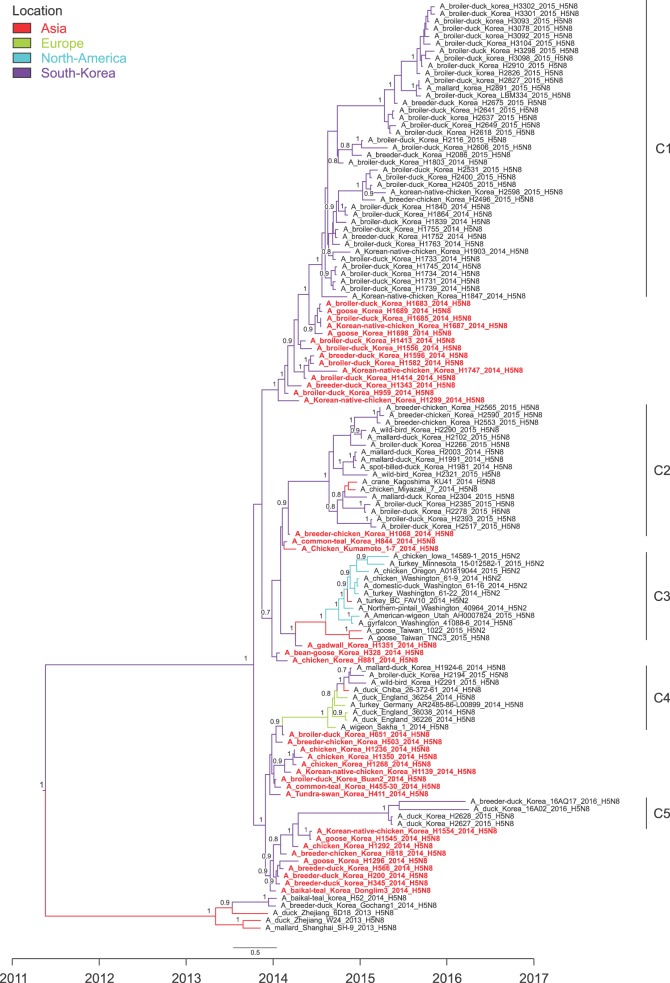 | Fig. 2The estimated maximum clade credibility (MCC) phylogeny of the hemagglutination (HA) gene of H5N8 in Korea and of H5 worldwide (all variants of H5 clade 2.3.4.4). Each subgroup is marked on the right and the C0 group is labeled in red. Branch lengths represent time, and colors represent location. Red, Asia; green, Europe; blue, North America; and purple, South Korea. Numbers at the nodes show posterior probabilities >0.7.
|
The C2 and C4 subgroups were reintroduced by wild birds at the end of 2014. These groups were included among the H5N8 viruses that spread to Europe, Japan, and Taiwan after further evolution in the breeding sites of migratory birds [2910]. The viruses were also introduced into poultry farms and circulated for 4–5 months in South Korea (panel B in Fig. 1). Although wild birds introduced HPAI from overseas countries, the spread of the viruses among poultry farms is generally the result of human activities, such as trade in infected poultry and mechanical movement of infected materials [1]. Over the 2014–2016 H5N8 outbreak period, reassortment viruses such as H5N2 or H5N3 HPAI were not detected (data not shown); however, they were detected in North America and Taiwan [611]. The internal genes of H5N8 viruses show only the accumulation of point mutations (range, 97.5%–100%; data not shown), which are observed in long-term outbreaks.
Preventing the introduction of HPAI viruses may be difficult in Korea because of geographical risk factors such as the presence of wintering sites for migratory birds and the close proximity to HPAI endemic countries. The 2014 to 2016 H5N8 HPAI outbreak was longer than the previous H5N1 HPAI outbreaks. This may be due to complex factors such as the characteristics of the H5N8 virus in the domestic duck, which excretes abundant amounts of virus without evident clinical signs, the high density of duck farms with low biosafety levels, and the vulnerability of detection systems in live bird markets or minor poultry farms. Furthermore, reintroduction of viruses by wild birds has played a key role in the long persistence of the H5N8 virus after the initial outbreak. Therefore, enhancing the biosafety level of farms, educating field employees, and enforcing facility regulations is essential in controlling HPAI viruses. Active surveillance of minor and major poultry farms, live bird markets, and wild birds is also of great importance.
The present study described the geographical and chronological distribution of H5N8 viruses in South Korea via a molecular epidemiological analysis of the relationships between HPAI outbreaks in wild birds and poultry. The present study may be useful in elucidating the epidemiology of H5N8 outbreaks in South Korea.
 XML Download
XML Download