

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Porcine epidemic diarrhea virus (PEDV) is a major cause of aqueous diarrhea, vomiting, and severe dehydration in pigs. It is transmitted via direct or indirect fecal-oral routes [1014]. As other Coronaviridae members, PEDV is an enveloped, positive-sense, single-stranded RNA virus with a small 28 kb genome. The genomic RNA has a 5′-cap and a 3′-polyadenylated tail and contains at least seven open reading frames that encode four structural proteins: spike (S), envelope (E), membrane (M), and nucleocapsid (N) [9]. Like other coronaviruses, the S protein is a type 1 transmembrane envelope glycoprotein comprising 1,383 amino acids and contains 29 predicted glycosylation sites. The S1 and S2 domains of this protein have essential roles in cellular receptor interactions that mediate viral entry and induce neutralizing antibodies in the natural host [13]. The PEDV S protein consists of three domains: a large extracellular domain, a transmembrane region, and a short cytoplasmic carboxyl terminus. There is a region of the S protein that contains the epitope(s), which is capable of inducing PEDV-neutralizing antibodies [1]. Research indicates that phage-displayed peptides, which were panned against 2C10, have antigenic similarities with the PEDV antigen epitope motif (1368GPRLQPY1374) and can show neutralizing activity against PEDV [6], the CO-26K equivalent (COE) domain (aa positions 99–638), and the epitopes SS2 (aa positions 748–755) and SS6 (aa positions 764–771), which can also induce neutralizing antibodies against PEDV [425]. Recently, the S gene was implicated as an important determinant of the biological properties of PEDV. These properties include genetic relationships between PEDV isolates, the epidemiological status of PEDV in the field, and associations between genetic mutations and virus function [16].
Porcine epidemic diarrhea (PED) was first reported in the United Kingdom in 1971, although the virus was not identified until 1978 when it was designated PEDV with the prototype strain CV777 [11]. Subsequently, PEDV infections have been reported in China, Japan, Korea, and Thailand and for the first time in the United States in 2013 [4,17,22,24]. In China, the first occurrence of PED was in 1973, although the causative agent was not identified until 1984 [27]. Since the end of 2010, PEDV infections have been detected in more than 10 provinces in southern China.
According to the previous study, there was a high prevalence rate of PED in pig herds [3]. Clinical signs of PED are seen in piglets within 7 days of birth and present as vomiting and aqueous diarrhea. Death rates during this stage range from 80% to 100% resulting in huge economic losses in the pig industry [17]. During the period from 2010 to 2013, the incidence of diarrhea among newborn piglets from 60 large-scale farms in Guangdong and some other southern provinces was 59,320, of which 45,601 piglets died, for an average mortality rate of 76.87%. These studies indicate that PED is an increasingly serious problem in China.
In this study, we sequenced and analyzed the S gene of 55 PEDV strains from South China. The aim was to provide an improved understanding of the origin, evolution, and diversity of the strains in this region, and to determine the phylogenetic relationships between newly isolated and historic strains.
Go to : 
Materials and Methods
Sample collection and processing
Between February 2011 and March 2015 in Guangdong province, China, porcine intestinal and fecal samples were collected from piglets with watery diarrhea and dehydration. The intestinal and fecal samples were placed in phosphate buffer saline supplemented with penicillin G (10,000 IU/mL) and streptomycin (2 mg/mL) and centrifuged at 1,500 × g for 15 min. Viral presence in the supernatant was determined by using RT-PCR with viral gene-specific primers: (5′ to 3′) GCAACTCAAGTGTTCTCAG and GAGTCATAAAAGAAACGTCCG. RNA extraction and reverse transcription were carried out as previously described [27]. PCR products were purified by using an AxyPrep DNA gel extraction kit (Axygen, USA) according to the manufacturer's instructions, and the product was cloned into the pMD-19T vector (Takara Bio, China). The purified recombinant plasmids were sequenced by Invitrogen Trading (Invitrogen, China). Sequences of all samples detected to be positive for the complete S gene of PEDV were submitted to GenBank (National Center for Biotechnology Information, USA) under accession Nos. KP399601–KP399634 and accession Nos. KR296663–KR296683.
Molecular analysis
Nucleotide and deduced amino acid sequences were aligned, edited, and analyzed with Clustal X (ver. 1.83) [5], BioEdit (ver. 7.0.5.2), and DNASTAR software. The phylogenetic tree was constructed by using MEGA (ver. 5.05) [26] incorporating the neighbor-joining (NJ) method. Bootstrap values were estimated for 1,000 replicates.
Go to :
Results
Sequence analysis of the S gene
Sequence analysis revealed that the S genes of the 55 Chinese PEDV strains ranged from 4149 to 4176 nucleotides in length. The S gene of strain CH-WTC1-02-2013 comprised 4149 nt, which is the same length as the Korean strain DR13. The S genes of CH-STC-12-2011, CH-XLC-03-2013, CH-SHC-12-2014, CH-HFEC-01-2015, CH-JPYC-02-2015, CH-XBC-01-2015, CH-YGC-01-2015, CH-ZWBZa-01-2015, and CH-ZWC-01-2015 consisted of 4152 nt and were the same length as those of the classic strains (including CV777, Br1-87, attenuated DR13, LZC, NL/GD001/2014, FR/001/2014, GER/L00719/2014, 25-10_2015_AUT, 15V010/BEL/2015, and USA/OH851/2014). The S region nucleotide sequences of CH-TPC-03-2013, CH-SHC-12-2013, CH-HGCP-12-2013, and CH-CCC-05-2013 were 4158 nt long, the same length as Chinese strains CHGD-01, GDS01, LC, and GXNN/2013. However, the S genes of CH-LNC-12-2012 and CH-LNC-10-2014 comprised 4176 nt, which is the same length as that of Japanese strain NK, and encoded a 1391 amino acid long peptide. The S genes of the other Chinese PEDV strains were 4161 nt long and were the same length as three Korean strains (KNU-0905, KNU-0801, and CNU-091222-01) and two USA strains (USA/Texas128/2014 and USA/Illinois260/2014).
Compared to the S gene of CV777, sequencing of the field strains revealed a range of lengths: one was three nt shorter (4149 nt), nine were the same length (4152 nt), four were six nt longer (4158 nt), 39 were nine nt longer (4161 nt) and two were 24 nt longer (4,176 nt).
The deduced amino acid sequences of the 55 S proteins ranged from 1382 to 1391 aa in length. While the SS2 epitope was conserved in all 55 Chinese PEDV strains, most strains exhibited two or three amino acid mutations in the SS6 epitope (Fig. 1). Among the 55 South China field isolates, 53 strains (i.e., excluding CH-YGC-11-2013 and CH-XLC-03-2013) had the same mutation (Y/F766S) in SS6. We also found that 52 strains (i.e., excluding CH-YGC-11-2013, CH-TPC-03-2013, and CH-CCC-05-2013) had the same mutations (G/S1368S, L/F1371S, and L/X1371S) in the 2C10 region (Fig. 1).
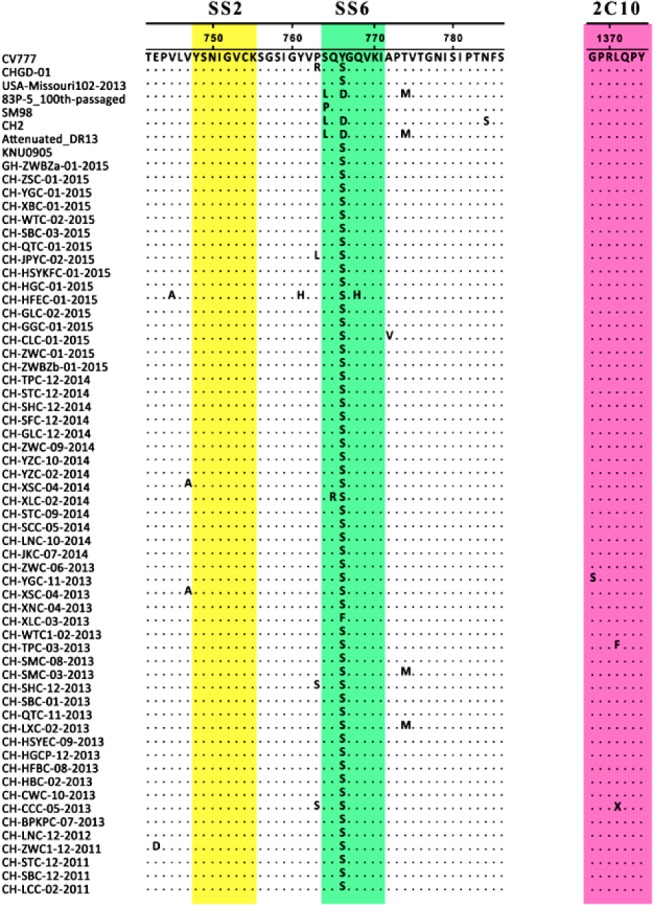 | Fig. 1Alignment of the deduced amino acid sequences of partial spike (S) protein of Guangdong isolates with that of vaccine strains. The dots represent amino acids that are identical to those in the CV777 strain. Boxes indicate the neutralizing epitopes (SS2, 748–755; SS6, 764–771; and 2C10, 1368–1374).
|
Phylogenetic analysis of the S gene
Phylogenetic analysis of S gene showed that all PEDV strain sequences could be divided into four groups (Fig. 2). One group (G1a) comprised the CV777, DR13, SM98, LZC, Br1-87, MK, Attenuated_DR13, 83P-5_100th-passaged and GDS03. The second group (G1b) consisted of CH2, CH5, CH6, CH7, JS-2004-2, LJB-03, DX from China, one USA strain (USA/OH851/2014), and strains recently isolated in Europe (NL/GD001/2014, FR/001/2014, GER/L00719/2014, 25-10_2015_ AUT, and 15V010/BEL/2015). Ten of the fifty-five South China PEDV strains belonged to G1b. The third group (G2a) was made up of three Korean strains (CNU-091222-01, KNU-0905, and Chinju99) and two Japanese strains (NK and KH). The last group (G2b) included 12 Chinese strains (CHGD-01, GDS01, LC, HuN, CH-AY-11, HB-2011-1, BJ-2011-2, CH-JLCC-2011, GDQY/2011, GDDG/2011, GXNN/2013 and GDZQ/2012), two Korean strains (KPV1406 and CNU-091222-01), three USA strains (USA/Texas128/2014, USA/Illinois260/2014, and USA/Missouri102/2013), and one Canadian strain (MB-021). Forty-five of the fifty-five South China PEDV strains belonged to G2b.
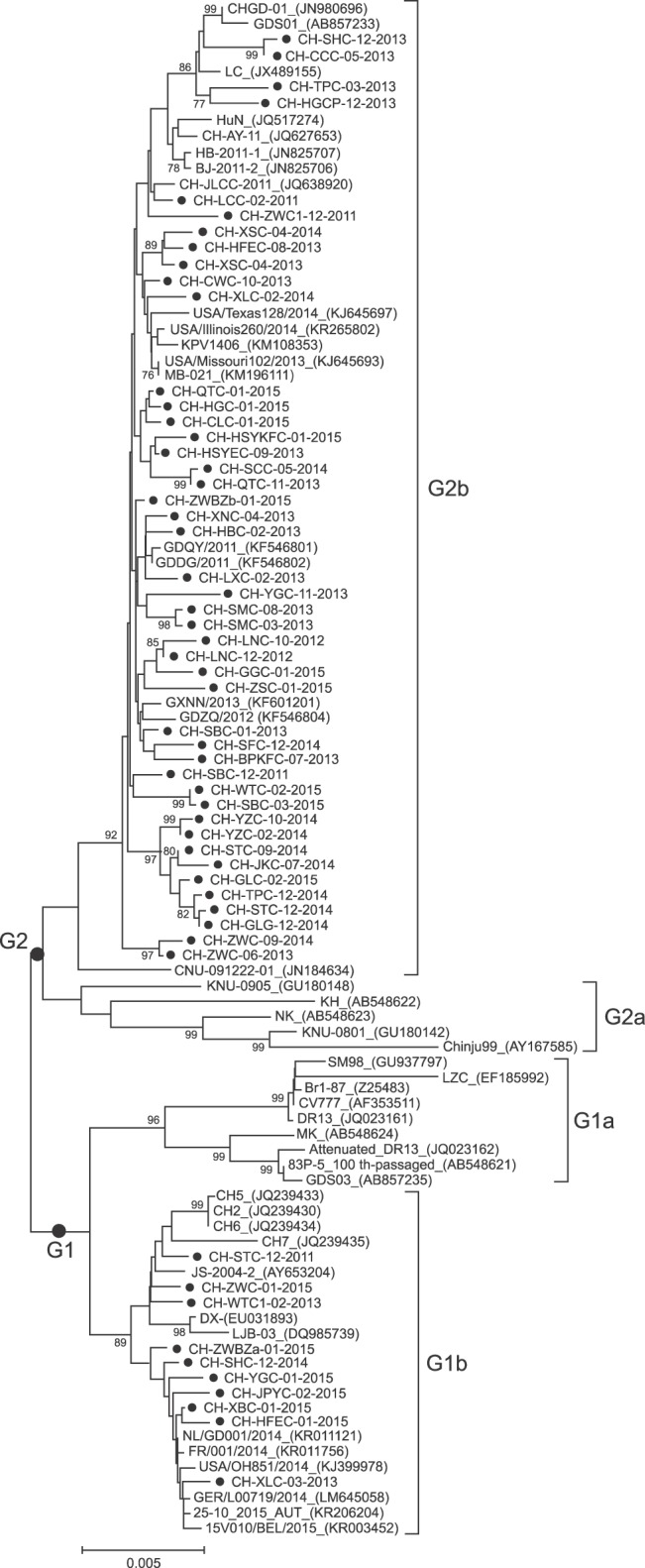 | Fig. 2Phylogenetic analysis of porcine epidemic diarrhea virus (PEDV) strains based on nucleotide sequences of the complete spike glycoprotein S gene. The tree was constructed using the neighbor-joining method in MEGA 5.05 software, using 1,000 bootstrap replicates. Bootstrap values > 75% are shown at the branch points.
|
Sequence identity analysis of the S gene
Sequence alignment showed nucleotide (deduced amino acid) identities in the range 94.4% to 99.9% (93.8%–99.9%) among the 55 Chinese PEDV strains and identities in the range 92.4% to 99.76% (90.7%–99.5%) with the foreign reference strains reported in GenBank. Some vaccine strains including CV777, attenuated DR13, SM98, and 83P-5_100th-passaged are used in a variety of countries. The S genes of our 55 recently isolated Chinese field strains shared identity with CV777 (93.8%–95.7%), CHGD-01 (94.6.0%–98.3%), SM98 (93.3%–95.4%), attenuated DR13 (93.3%–95.6%), and 83P-5_100th-passaged (93.7%–96.0%). The deduced amino acid sequences of these strains shared identity with CV777 (93.0%–96.1%), CHGD-01 (94.1%–98.3%), SM98 (92.1%–95.3%), attenuated DR13 (92.0%–95.7%), and 83P-5_100th-passaged (92.4%–96.1%). The S genes of the 55 recently isolated Chinese field strains shared nucleotide (deduced amino acid) homologies of 96.1%–99.2% (95.1%–99.5%) with USA/Missouri102/2013.
Prediction of N-glycosylation
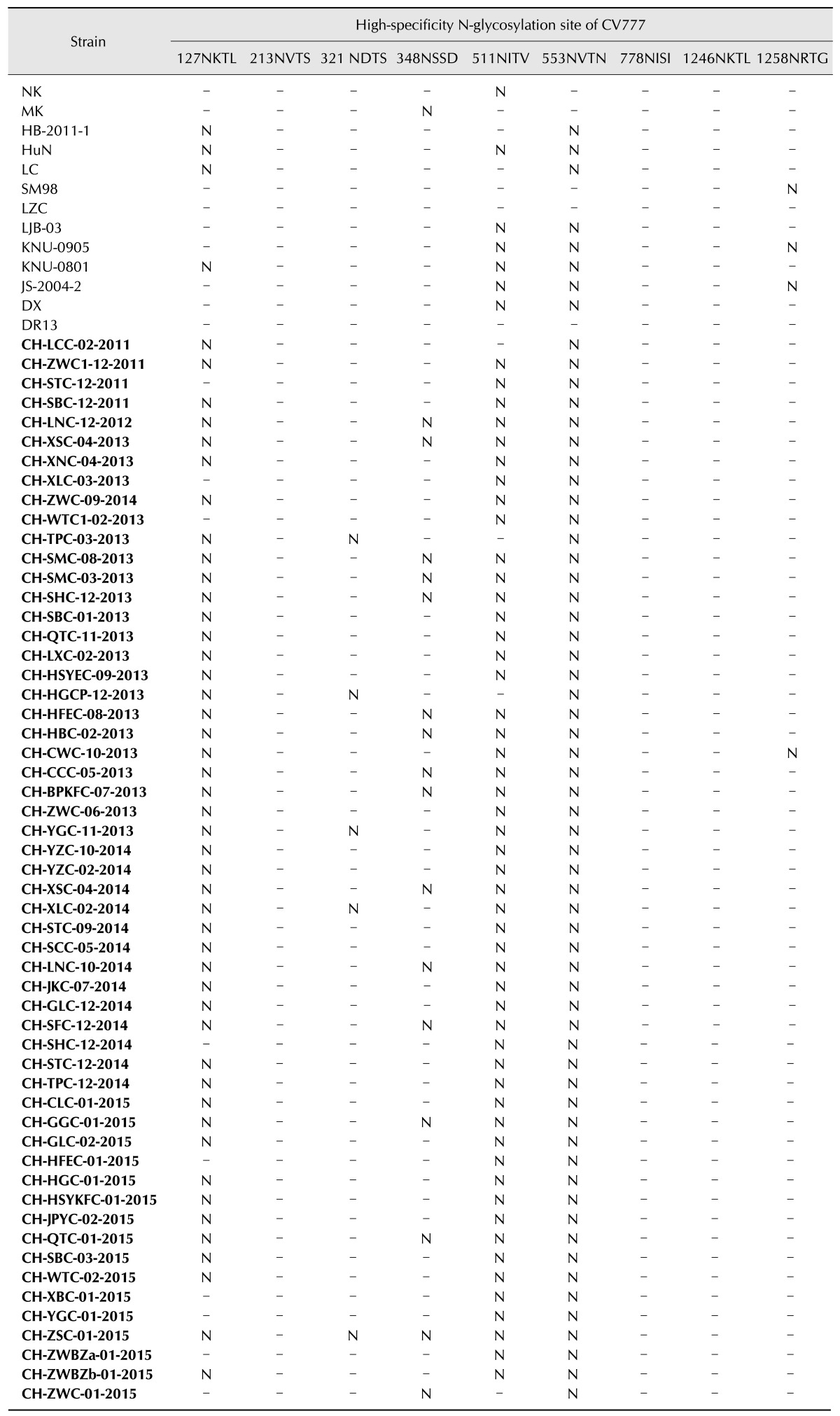
All of the analyzed strains contained between nine and ten high-specificity N-glycosylation sites, three of which were conserved. The CV777 strain is the only vaccine source in China. Therefore, a comparative analysis of all 55 strains and CV777 was conducted. All of the Chinese field strains, except CH-SHC-12-2014, CH-HFEC-01-2015, CH-XBC-01-2015, CH-YGC-01-2015, CH-ZWBZa-01-2015, CH-ZWC-01-2015, CH-XLC-03-2013, CH-WTC1-02-2013, and CH-STC-12-2011, lost the Asn-Xaa-Ser/Thr-127NKTL high-specificity N-glycosylation sites, which are conserved in vaccine strains (including CV777) (Table 1). One additional N-glycosylation site Asn-Xaa-Ser/Thr-62NSTW, which was not occur in the CV777 strain, presented in CHGD-01, KNU-0801, KNU-0905, and all of the Chinese field isolates except CH-SHC-12-2014, CH-HFEC-01-2015, CH-XBC-01-2015, CH-YGC-01-2015, CH-ZWBZa-01-2015, CH-ZWC-01-2015, CH-XLC-03-2013, CH-WTC1-02-2013, and CH-STC-12-2011. Within the South China sequence set, 52 strains (i.e., excluding CH-XLC-03-2013, CH-WTC1-02-2013, and CH-STC-12-2011) had similar N-glycosylation conformations. Fifty-one strains (i.e., strains excluding CH-ZWC-01-2015, CH-HGCP-12-2013, CH-TPC-03-2013, and CH-LCC-02-2011) had lost the Asn-Xaa-Ser/Thr -511NITV site, which was contained in the other South China strains (Table 1).
Go to :
Discussion
This study was carried out to elucidate the diversity of PEDV in South China. We determined the phylogenetic relationships between new and historic PEDV strains in this region and compared them with globally reported strains. To that end, we sequenced and analyzed the S glycoprotein gene of 55 Chinese field strains isolated from South China during 2011–2015.
All 55 Chinese PEDV strains had a high sequence similarity with the previously identified Guangdong strains (CHGD-01, GDQY/2011, GDZQ/2012, GXNN/2013, and GDS01). The results further indicate the homogeneity in the S genes of PEDV field strains prevalent in South China. Our results also demonstrate that the recently identified Guangdong PEDV strains are closely related to those isolated from the USA, indicating that the USA strains may have originated from Chinese variants [12].
High morbidity and mortality were observed in pigs that were immunized with inactivated or attenuated PED vaccines based on CV777 [19], suggesting that the CV777 vaccine is unable to provide full protection in swine. We have reported on further investigations into the S gene mutations responsible for the failure or otherwise of the existing CV777 vaccine [28].
The S genes of attenuated strains (CV777-attenuated, attenuated DR13, and 83P-5_100th-passaged) are 4149 nt in length, while the classical virulent strains, such as CV777, are 4152 nt long [1618]. Sequence analysis revealed length diversity among the 55 Chinese field strains in this study. The S gene of one field strain was shorter than those of the classical strains due to nucleotide deletions, while the S genes of 45 field strains were longer due to nucleotide insertion. These variations are indicative of the unique characteristics of the S genes of the 55 field strains in the present study.
With the exception of the CH-YGC-11-2013 and CH-XLC-03-2013 strains, the remaining 53 South China strains contained several amino acid substitutions compared with the vaccine strains, resulting in changes in the amino acid constitution of the SS6, 2C10, and COE neutralizing epitopes. A retrospective study of the three amino acid substitutions revealed that the shift process was present not only in European strains but also in Asian PEDV strains [8]. Those results in combination with our results indicate that the three substitutions represent a marker of increased PEDV viability [8]. The domestic outbreak of PED in China was caused by new PEDV strains, the S genes of which had similar molecular characteristics [7]. According to previous studies as well as the present study, the epidemic strains of PEDV have a close genetic relationship with Asian strains including those isolated in China, Korea, and Japan after 2010 [2,15,20]. Therefore, a detailed description of the differences between epidemic strains and vaccine strains could aid in the development of improved vaccines against PEDV.
Previous research has shown that the Severe Acute Respiratory Syndromes (SARS) corona virus (CoV) originated from Guangdong Province in southern China and has a recombinant history with lineages of type I coronavirus such as PEDV [21]. Moreover, the high frequency of recombination events in CoVs may result in the generation of novel viruses with high genetic diversity, and it could invoke unpredictable changes in virulence during human infections [23]. As such, the information we have gathered about the mutation of S genes could be informative for monitoring and predicting the potential emergence of a recombinant CoV from animals.
In summary, our sequence analysis of the S genes and molecular epidemiology of 55 field strains from South China indicate that the PEDVs circulating between 2011 and 2015 in this region have evolved from historic strains within the local region. The results of our analyses will be useful when improving the PED control strategies in this region.
Go to :
 XML Download
XML Download