

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Temporal lobe epilepsy, the most prevalent form of focal epilepsy in adults, is associated with hippocampal sclerosis, which is characterized by neuroanatomical and neuropathological alterations such as neuronal loss and gliosis in the hippocampus [18]. Rat or mouse models of status epilepticus (SE) induced by systemic administration of pilocarpine have been extensively utilized to study human temporal lobe epilepsy [2410]. Pilocarpine-induced SE is known to cause extensive neuronal damage in the mouse hippocampus [2627]. The underlying mechanisms associated with neuronal death in the SE have been suggested to include glial activation, neuroinflammation, oxidative stress, glutamate receptor mediated-excitotoxicity, and high calcium influx in neurons [21242829]. However, these mechanisms do not fully explain neuronal damage following SE.
Hippocalcin, which contains four calcium-binding sites and an amino-terminal myristoylation site, belongs to the neuronal calcium sensor and participates in the maintenance of calcium homeostasis [311]. Hippocalcin is most abundant in the hippocampus among various brain regions, and plays important roles in postsynaptic neural functions and synaptic plasticity [22]. Hippocalcin can also bind the adaptor protein 2 complex and regulate the hippocampal long-term depression [18]. Hippocalcin acts as a molecular linker between calcium entry through N-methyl-D-aspartate receptors into neurons and the regulated endocytosis of Ν-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors [18]. Moreover, hippocalcin gates the calcium-dependent potassium channels, which are related to the slow afterhyperpolarization in hippocampal pyramidal cells [26]. Hippocalcin is also known to interact with neuronal apoptosis inhibitor protein, and to be involved in neuroprotection against calcium-induced cell death [17]. Hippocalcin level was also recently reported to be decreased in the rat cerebral cortex following focal cerebral ischemia, and glutamate toxicity decreased hippocalcin level in cultured mouse hippocampal cell lines [12]. However, the changes in hippocalcin level in the hippocampus following epilepsy have not yet been fully elucidated. Therefore, in this study, we examined the time-course changes of the hippocalcin protein level in the mouse hippocampus following pilocarpine-induced SE.
Materials and Methods
Experimental animals
Eight-week-old male ICR mice were purchased from RaonBio (Yongin, South Korea). Animals were housed at adequate temperature (22 ± 3℃) and humidity (55 ± 5%) control under a 12-h light/12-h dark cycle while provided with free access to food and water. All experimental procedures for animal handling and use were approved by the Institutional Animal Care and Use Committee at Dankook University.
Induction of SE
For induction of SE, the animals were intraperitoneally (i.p.) treated with 300 mg/kg pilocarpine (Sigma-Aldrich, USA) 30 min after scopolamine butylbromide (1 mg/kg, i.p.; Sigma-Aldrich) to reduce the peripheral cholinergic effects. Pilocarpine and scopolamine were dissolved in saline. Animals began to show seizure activity within 20 to 30 min of treatment with pilocarpine. The seizure activity was monitored behaviorally and scored according to the Racine scale [20]. Only animals reaching at least stage 4 (rearing, myoclonic twitching) were included in this study. At 2 h after onset of SE, 5 mg/kg diazepam (Valium; Roche, France) was administered i.p. to terminate SE. Mice in the control group received the scopolamine and diazepam, but received saline instead of pilocarpine.
Tissue processing for histology
Mice in the control and pilocarpine-treated groups (n = 7 at each time point) were sacrificed at designated times (6, 12, 24 and 48 h after SE). For histological analysis, animals were anesthetized with Zoletil 50 (30 mg/kg; Virbac, France) and perfused transcardially with 4% paraformaldehyde in 0.1 M phosphate-buffer. The brain tissues were then removed and serially sectioned with a cryostat (Leica, Germany) into 30 µm coronal sections. Sections containing the hippocampus were used according to anatomical landmarks corresponding to bregma −1.70 to −2.46 mm of the mouse brain atlas [9].
Fluoro-Jade B staining
To examine neuronal damage in the hippocampus after SE, Fluoro-Jade B (F-J B) histofluorescence staining was performed as previously described [1523]. In brief, the sections were stained with solution containing 1% sodium hydroxide, a solution of 0.06% potassium permanganate and 0.0004% Fluoro-Jade B (Histochem, USA) staining solution. The sections were examined using an epifluorescent microscope (Carl Zeiss, Germany). F-J B positive (+) cells exhibiting bright green fluorescence and profiles of neuronal somas were counted on representative microscopic fields (20× magnification) as previously described [27], while F-J B+ fragments were not counted. F-J B+ cells were counted in the center of the hippocampal cornu ammonis 1 (CA1) and CA3 regions, in the CA2 region and in the polymorphic layer of dentate gyrus (DG) per section.
Immunohistochemistry for hippocalcin
As described in our previous study [15], immunohistochemical staining for hippocalcin was conducted using rabbit anti-hippocalcin (1 : 500, Abcam, USA), biotinylated goat anti-rabbit IgG (1 : 200, Vector, USA) and streptavidin peroxidase complex (1 : 200, Vector) and visualized with 3,3'-diaminobenzidine in 0.1 M Tris-HCl buffer.
Six sections with 90 µm intervals per animal were selected to quantitatively analyze hippocalcin immunoreactivity. Digital images of hippocampal subregions were captured with an AxioM2 light microscope (Carl Zeiss) using a digital camera (Axiocam; Carl Zeiss). Semi-quantification of the immunostaining intensities in the pyramidal cells of the hippocampal CA1-3 region and in the polymorphic and granular cells of the dentate gyrus were evaluated with Image J 1.46 (National Institutes of Health, USA) according to the method described in our previous study [15]. The mean intensity of immunostaining in each immunoreactive structure was measured using a 0 to 255 gray scale system, after which the level of immunoreactivity was scaled as −, ±, + or ++, representing no staining (gray scale value: ≥ 200), weakly positive (gray scale value: 150–199), moderate (gray scale value: 100–149) or strong (gray scale value: ≤ 99), respectively.
Western blot analysis for hippocalcin
To examine changes in hippocalcin protein levels in the hippocampus after SE, mice in the control and pilocarpine-treated groups (n = 7 at each time point) were used for western blot analysis at designated times (6, 12, 24 and 48 h after SE) as described in our previous study [15]. Briefly, the hippocampus was homogenized and centrifuged, after which the supernatants were subjected to western blot analysis. Rabbit anti-hippocalcin (1 : 1,000, Abcam) or mouse anti-β-actin (1 : 5,000; Sigma-Aldrich) was used as a primary antibody. The results of the western blot analysis were scanned and subjected to densitometric analysis for quantification of the bands using Image J 1.46 to determine the relative optical density (ROD). Hippocalcin level was normalized against the β-actin level. The ROD was reported as % with the control-group designated as 100 %.
Results
Neuronal degeneration after SE
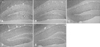
F-J B+ cells were not observed in the hippocampal subregions in the control group (Table 1; panel A in Fig. 1). At 6 h after SE, F-J B+ cells began to be detected in the mouse hippocampus, especially in the CA1 region and the DG (Table 1; panel B in Fig. 1). In the CA1 region, the number of F-J B+ cells increased in a time-dependent fashion, and marked neuronal degeneration was observed in most of the pyramidal neurons at 24 h and 48 h after SE (Table 1; panels C–E in Fig. 1). However, only some pyramidal neurons showed F-J B histofluorescence in the CA2 region at 24 h after SE (Table 1; panels C and D in Fig. 1). In the CA3 region, F-J B+ cells were detected from 12 h after SE, and were markedly increased in the most pyramidal neurons at 24 h and 48 h after SE (Table 1; panels C–E in Fig. 1). In the DG, neuronal degeneration began to be observed in polymorphic cells from 6 h after SE, and F-J B+ polymorphic cells were detected in high levels at 12 h and 24 h after SE (Table 1; panels B–E in Fig. 1).
Changes in hippocalcin immunoreactivity after SE
In the CA1 region of the control-group, intense hippocalcin immunoreactivity was detected in the pyramidal neurons of the stratum pyramidale (SP), as well as in the stratum oriens (SO) and stratum radiatum (SR) (Table 2; panel A in Figs. 2 and 3). However, almost no hippocalcin immunoreactivity was detected in the pyramidal neurons from 6 h until 48 h after SE (Table 2; panels B–E in Figs. 2 and 3). In addition, decreased hippocalcin immunoreactivity in the SO and SR was observed at 24 h and 48 h after SE (Table 2; panels D and E in Figs. 2 and 3).
In the control-group, hippocalcin immunoreactivity was detected in the pyramidal neurons of the SP as well as in the SO and SR in the CA2/3 region (Table 2; panel A in Figs. 2 and 4). Hippocalcin immunoreactivity in the SP was markedly decreased from 6 h after SE (Table 2; panel B in Figs. 2 and 4). In the CA3 region, only a few hippocalcin+ cells were observed at 12 h after SE, and almost no hippocalcin+ cells were detected at 24 h after SE (Table 2; panels C and D in Figs. 2 and 4). However, many pyramidal neurons in the CA2 region showed hippocalcin immunoreactivity until 24 h after SE (Table 2; panel D in Figs. 2 and 4). At 48 h after SE, no hippocalcin+ cells were not observed in the CA2/3 region (Table 2; panel E in Figs. 2 and 4).
In the DG of the control-group, hippocalcin immunoreactivity was observed in the granule cells of the granule cell layer and the polymorphic cells of the polymorphic layer (Table 2; panel A in Figs. 2 and 5). Hippocalcin immunoreactivity in the polymorphic cells was markedly decreased at 6 h after SE, and almost no immunoreactivity was detected from 12 h until 48 h after SE (Table 2; panels B–E in Figs. 2 and 5). In addition, hippocalcin immunoreactivity in the granule cell layer was decreased at 24 h and 48 h after SE (Table 2; panels D and E in Figs. 2 and 5).
Changes in hippocalcin protein level after SE
Western blot analysis revealed that the pattern of time-course changes in hippocalcin protein levels in the mouse hippocampus following pilocarpine-induced SE was generally similar to that observed in the immunohistochemical data. Hippocalcin protein level in the hippocampus began to decrease from 6 h after SE, after which hippocalcin protein level was significantly decreased at 24 h and 48 h following pilocarpine-induced SE (Fig. 6).
Discussion
Epilepsy, which is characterized by spontaneous recurrent seizure, occurs via paroxysmal alterations in neurological functions caused by abnormal electrical activity in the brain [5]. In addition, it is well known that brain dysfunctions such as transient and abnormal hyperactivity in neuronal populations of the brain are accompanied by motor and sensory disturbances, and that seizure results from excessively enhanced neuronal excitation or deficient neuronal inhibition [14].
Previous studies have shown that pilocarpine led to seizure via activation of the muscarinic acetylcholine receptor, and that neuronal degeneration in the rat hippocampus was highly extended from 1 to 7 days after pilocarpine administration [719]. In the present study, we observed neuronal degeneration in the CA1 region and DG at 6 h after SE and in the CA2/3 region at 12 h after SE, respectively. We also observed that neuronal degeneration increased in a time-dependent manner, and that neuronal degeneration occurred in most of the pyramidal neurons of the mouse hippocampus at 24 h and 48 h after pilocarpine-induced SE. These findings were consistent with those of previous studies that showed the evolution of neuronal damage in mice after pilocarpine administration [627]. It has been reported that neuronal degeneration in the mouse hippocampus occurred at 4 h after onset of SE [27]. Specifically, that study showed that neuronal degeneration reached peak levels at 3 to 5 days in the hippocampal CA1-3 region and at 4 h in the polymorphic layer of the DG. Neuronal damage is also known to be triggered by pilocarpine-induced SE, and most polymorphic cells undergo neuronal degeneration as early as 6 h after SE [2]. Moreover, the maximum degree of neuronal damage in the polymorphic layer was seen as early as 3 h after SE, and the greatest neuronal damage was observed in the CA1 and CA3 pyramidal cell layers at 1 week after SE [6]. Therefore, based on the results of the present and previous studies, it is likely that pilocarpine-induced SE leads to the massive neuronal degeneration in the mice hippocampus.
It is well known that the regulation of intracellular calcium concentration by intracellular calcium-binding proteins is important to maintenance of cellular homeostasis [25]. A sustained increase of intracellular calcium concentration in neurons during and/or following seizure has also been reported to lead to neuronal death [29]. Furthermore, it is widely accepted that a sustained elevation of intracellular calcium concentration is one of the most important mediators of excitotoxic neuronal damage in epilepsy [28]. Therefore, failed regulation of intracellular calcium concentration is thought to be associated with the pathogenesis of epilepsy. In the present study, we found that hippocalcin immunoreactivity and protein level decreased from 6 h after SE, and that low hippocalcin immunoreactivity and protein level were observed at 24 h and 48 h after SE. A previous study showed that seizure-induced neuronal death was enhanced in the CA3 region of hippocalcin-knockout mice compared to wild-type mice after intraperitoneal injection of kainic acid [13]. In addition, the lack of hippocalcin in hippocalcin-knockout mice was reported to cause hippocampal neurons to be more vulnerable to damage by quinolinic acid through NMDA receptor stimulation, and the lack of hippocalcin might lead to slow calcium extrusion [16]. These findings suggest that hippocalcin may protect hippocampal neurons against calcium-dependent excitotoxin damage by enhancing calcium extrusion [16]. Therefore, it can be postulated that SE-induced decreases in hippocalcin may be closely associated with neuronal degeneration in the mouse hippocampus following pilocarpine-induced SE.
However, in the present study, we found that there was a difference in the reductions of hippocalcin immunoreactivity according to the hippocampal subregions. Almost no hippocalcin immunoreactivity in the pyramidal neurons of the CA1 region or in the polymorphic cells of the DG were detected 6 h after SE, whereas a marked SE-induced reduction of hippocalcin immunoreactivity in the pyramidal neurons of the CA3 region was observed 12 h after SE. Hippocalcin mRNA was reported to be expressed most intensely in the CA3 region among the hippocampal subregions [22]. Therefore, it can be assumed that differences in SE-induced neuronal degeneration among hippocampal subregions are due to the abundance of hippocalcin levels. In addition, in this study, we observed that many pyramidal neurons in the CA2 region represented hippocalcin immunoreactivity until 24 h after SE, and that there was less neuronal degeneration in the CA2 region than in other hippocampal subregions. Although the exact mechanism of this phenomenon is unclear, it can be concluded that pyramidal neurons in the CA2 region are more resistant against SE-induced neuronal damage and that the activity of hippocalcin lasts much longer in the CA2 region than in the CA1 and CA3 regions following pilocarpine-induced SE.
In summary, we observed that both neuronal degeneration and decreased hippocalcin levels were detected in the mouse hippocampus after SE, although there were differences among hippocampal subregions. These findings indicate that the marked reduction of hippocalcin level may be closely related with the neuronal degeneration in the hippocampus following pilocarpine-induced SE.







 XML Download
XML Download