

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Stroke is a leading cause of neurological disability and the second most frequent cause of mortality in humans worldwide [3]. Ischemic stroke accounts for approximately 87% of all stroke cases while intracerebral hemorrhage accounts for 10% and subarachnoid hemorrhage accounts for 3%. A frequent cause of ischemic stroke is embolic or thrombotic occlusion of a major cerebral artery, most commonly the middle cerebral artery (MCA) [3].
The complexity of stroke, including clinical variability caused by differences in the duration, location, and severity of ischemia as well as the patient's age and concomitant diseases complicates clinical research on this condition [3]. In addition, complexity of the brain and response of this organ to stroke make it impossible to recapitulate stroke and its consequences using in vitro systems alone [3]. For these reasons, appropriate animal models must be developed to elucidate the complex pathophysiology of stroke, and find effective preventative and therapeutic approaches [3].
Since the 1970s, various animal models of cerebral ischemic stroke have been established to investigate the mechanisms of ischemic damage during ischemic stroke and possible therapeutic modalities [6,37]. Focal and global ischemia models have been most widely studied using a variety of methods. Global ischemia results from a reduction of blood flow affecting the entire brain whereas focal cerebral ischemia is produced by selective reduction of cerebral blood flow in a certain area. The great advantage of focal cerebral ischemia models is that selective blood vessel occlusion most commonly occurs in the middle cerebral artery (MCA) and mimics the pathophysiology of ischemic stroke in humans [3,14].
Mechanical occlusion of the MCA is accomplished by two main methods: proximal MCA occlusion (pMCAO) and distal MCA occlusion (dMCAO). pMCAO is most frequently used in animal models of ischemic stroke and is generally induced by direct mechanical occlusion, most commonly via advancing a silicon-coated suture through the internal carotid artery to the origin of the MCA in the circle of Willis. Severity of ischemic brain injury can be adjusted (i.e., transient vs. permanent) by changing the time during which the silicon suture is left in place. Induction of ischemic stroke by dMCAO is performed via craniotomy to directly expose and handle the superficial branches of the MCA, and can therefore produce side effects including subarachnoid hemorrhage and cerebral infection [3,7]. In contrast, pMCAO does not require craniotomy and has additional advantages such as high reproducibility and similarity of the resulting focal cerebral ischemic damage to that of stroke in humans [20,21]. For these reasons, pMCAO has been the most frequently used in previous animal model studies.
The animals reportedly used as cerebral ischemic models to date can be divided into two main size-based categories [14]: rodent models (e.g., mice [7], rats [20], and gerbils [37]) and non-rodent models (e.g., dogs [14], cats [10], pigs [40], sheep [5], and non-human primates [6]). Non-rodent models have several obvious advantages over rodent models. First, various samples, including cerebrospinal fluid (CSF) and blood, can be obtained serially from the same animal. Second, gyrencephalic brains of these species closely resemble the human brain both structurally and functionally. Finally, the larger brain size of non-rodent animals provides optimal resolution for imaging techniques such as magnetic resonance imaging (MRI) [14]. Out of all non-rodent models, non-human primates most closely resemble humans but their use in such research involves additional ethical, economic, housing, and handling problems [14]. For these reasons, dogs can be a useful alternative for studying ischemic stroke [11,14].
The pathophysiology of stroke is complex and involves diverse mechanisms including cytokine-mediated inflammation, energy failure, loss of cellular ion homeostasis, acidosis, increased intracellular calcium levels, excitotoxicity, free radical-mediated damage, generation of arachidonic acid products, complement activation, disruption of the blood-brain barrier (BBB), glial cell activation, and leukocyte infiltration [16]. Among these factors, cytokine-mediated inflammation has been increasingly considered to be an important contributor to the pathophysiology of stroke [16,17]. Microglia and astrocytes are activated after ischemic insult and release various inflammatory mediators, including numerous cytokines. The expression of cell adhesion molecules on vascular endothelial cells is upregulated concomitantly, allowing infiltration of peripherally derived inflammatory cells into the ischemic area. These inflammatory cells also secrete additional cytokines that further activate microglia and astrocytes. All of these processes can result in further neuronal cell death and exacerbate the ischemic damage [16].
Three potent inflammatory cytokines that play critical roles in the pathophysiology of ischemic stroke are tumor necrosis factor alpha (TNF-α), interleukin (IL)-1, and IL-6 [7,12,17,26,30,31,33,34,39]. Previous reports [7,12,30,31,33] concerning experimental ischemic stroke (focal cerebral ischemia) in rodents have indicated that these cytokines can modulate the extent of ischemic damage. These results are supported by several lines of evidence, such as the production of TNF, IL-1, and IL-6 in rodent brains with ischemic stroke by resident microglia, intrathecal macrophages, infiltrating macrophages, monocyte-derived macrophages, and neurons [24,30,31]. The effects of TNF-α [12,26,39], IL-1 [12,38], and IL-6 [12,30] on neurons during experimental cerebral ischemia have been studied in detail.
Based on various lines of evidence including results obtained from experimental ischemic stroke models, associations between these inflammatory cytokines and clinical prognosis have been evaluated in several clinical studies of human ischemic stroke. Cytokine levels in peripheral blood samples have been most frequently measured [32,38] because of the relative ease of sampling although cytokines in CSF were also evaluated [33,34]. Comparatively few reports have assessed brain cytokine levels post-mortem due to difficulties with obtaining brain tissues from patients who did not survive ischemic stroke [26].
Overall, findings from existing studies of cytokines in cases of stroke, including those performed with experimental ischemic stroke models and clinical investigations of human cerebral ischemia, suggest that cytokines play a critical role in determining the severity, lesion size, and prognosis of ischemic stroke patients. In addition, the only currently approved pharmacologic intervention for ischemic stroke is recanalization of the affected vessel by recombinant tissue plasminogen activator. This procedure is associated with several risks including additional reperfusion injury and hemorrhage. Additional neuroprotective strategies are therefore needed. Inhibition or blockage of cytokine responses could be alternative therapeutic strategies. The purpose of the present study was to experimentally induce ischemic stroke in dogs through permanent MCAO and evaluate post-ischemic changes in clinical, MRI, histopathological, and immunohistochemical data over time. In addition, associations of CSF and tissue levels of IL-6 with clinical prognosis were analyzed.
Materials and Methods
Experimental animals
Nine healthy beagle dogs (mean body weight, 11.7 kg; range, 9.0~14.5 kg; 5 females and 4 males; mean age, 3.2 years; range, 2~4 years) were used for this study. The dogs were monitored for at least 2 weeks before the surgical procedure. Results of the physical and neurological examinations during this period were consistently normal. The animals were screened for metabolic diseases by complete blood count (CBC) and serum chemistry analysis, and were also screened for infectious pathogens such as canine parvovirus (CPV) and canine distemper virus (CDV). Each dog was housed in an individual cage and fed commercial dry food (Limited Ingredient Diets; Natural Balance Pet Foods, USA) twice daily. The end points of the study were 1 week and 2 weeks following induction of ischemic stroke via permanent MCAO. All dogs were euthanized at these end points except those that died suddenly due to severe neurological deterioration. The surgical and experimental protocols, including euthanasia, were approved by the institutional animal care and use committee (IACUC) of Gyeongsang National University (Korea).
Embolus preparation
Artificial silicon emboli were prepared as previously described [14]. Briefly, a silk suture (4-0 Black Silk; Ailee, Korea) was passed into the tip of a 20-gauge venous catheter (BD Angiocath Plus; Becton, Dickinson and Company, USA) and looped at the hub before passing back out of the tip. Artificial silicon material was prepared by mixing silicon rubber (90%) and curing agent (10%; SILASTIC MDX4-4210; Dow Corning, USA). The silicon material was then injected into the silk suture-containing catheter and allowed to cure for 24 h. After curing, the catheter was removed from the silicon-embedded suture. A 7-mm length of silicon-embedded suture was prepared for injection and inserted into the tip of a 30-mL syringe (Kovax-Syringe; Korea Vaccine, Korea) filled with physiologic saline.
Animal preparation, monitoring, surgical procedure, and recovery
All dogs fasted for more than 12 h before the induction of general anesthesia. The animals were premedicated with atropine (0.02 mg/kg SC, Jeil Pharm Atropine Sulfate Inj; Jeil Pharmaceutical, Korea) for 10 min, and anesthesia was induced with propofol (6 mg/kg IV, Provive; Myungmoon Pharm, Korea). The dogs were intubated orally and isoflurane (Terel Solution; Piramal Critical Care, USA) was administered at 2% to 3% of the inspired volume to maintain general anesthesia during the surgical procedures. The heart rate, body temperature, and blood oxygen saturation were monitored throughout the procedures.
Ischemic stroke was induced by MCAO as previously described [14]. The animal was placed in a lateral recumbent position. The cervical area was shaved, disinfected with betadine and alcohol scrubs, and incised to expose the common carotid artery. The carotid sheath was exposed by blunt dissection aided by palpation of the carotid pulse. The common carotid artery was separated from the vagosympathetic trunk, and the internal (a branch of the common carotid artery that supplies the anterior part of the brain) and external carotid arteries were identified. An 18-gauge venous catheter (BD Angiocath Plus; Becton, Dickinson and Company) was inserted directly into the internal carotid artery through the carotid bulb. After catheterization, a 30-mL syringe loaded with a silicon-embedded suture was mounted at the hub of the 18-gauge catheter. The silicon embolus was then flushed into the internal carotid artery and up to the origin of the MCA in the circle of Willis. Successful delivery of the silicon embolus was confirmed by monitoring for arterial "back flow" into the syringe. The 18-gauge catheter was removed and pressure was applied to the catheterization site to achieve hemostasis. After hemostasis was achieved, the cervical incision was closed with sutures (3-0 Black Silk; Ailee). The MCA was permanently occluded in all dogs until the end of the study. After surgery, tramadol (5 mg/kg IV, Humedix Tramadol Hcl Injection; Humedix, Korea) and ceftriaxone (30 mg/kg IV, Myungmoon Ceftriaxone Sodium Injection; Myungmoon Pharm) were administered for pain relief and as an antibiotic prophylaxis for 1 week. The incision site was disinfected daily with betadine ointment (Potadine; Samil Medical, Korea) and antibiotic ointment (Nitrofurazon Oint; Ahngook Pharm, Korea), and kept bandaged for 1 week.
Assessment of neurological status
Neurobehavioral scoring was performed using a standardized neurological system that has been previously described [14]. Neurological categories assessed were motor function (1, no deficit, ambulates normally; 2, hemiparetic but ambulates without assistance; 3, stands only with assistance; 4, hemiplegic, unable to stand, comatose, or deceased), level of consciousness (1, normal responsiveness; 2, mildly reduced responsiveness; 3, severely reduced responsiveness; 4, comatose or deceased), head turning (0, normal posturing of the head; 1, tends to turn head toward side of infarct; 2, unable to lift head, comatose, or deceased), circling (0, absent; 1, circling toward side of infarct, unable to test because comatose or deceased), and hemianopsia (0, normal vision; 1, clear-cut asymmetry, unable to test because comatose or dead). The total scores produced by this system ranged from 2 (completely normal) to 11 (most severe deficits, comatose, or deceased). Each dog was assessed prior to the induction of ischemic stroke and daily thereafter until death or euthanasia.
MRI
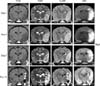
MRI was performed before and 1, 3, 7, and 14 days after induction of ischemic stroke using a 0.4-Tesla magnet MRI system (APERTO 0.4; Hitachi Medical Systems, Japan) to identify the location, extent, and progression of the infarcted lesion (s). All dogs were fasted for at least 12 h before each scan. Premedication and anesthesia were administered using the same protocol described for the surgical procedure. Sagittal and transverse T1-weighted images (T1WI), T2-weighted images (T2WI), fluid-attenuated inversion recovery (FLAIR) data, and diffusion-weighted images (DWI) were obtained under general anesthesia. Imaging software (Lucion ver. 1.50; Infinitt Health Care, Korea) was used to calculate ischemic lesion volumes based on the T2WI images for all dogs. Areas containing lesions with higher signal intensity in each slice of the T2WI images were summed to determine the total ischemic lesion volume. The volume of the whole brain was calculated in the same way for each dog. Lesion volume is expressed as the percentage of the whole brain volume occupied by ischemic lesions.
CSF analysis
CSF was collected from all dogs before and 1 h, 1 day, 3 days, 7 days, and 14 days after the induction of ischemic stroke except for cases in which the dog died acutely. CSF was obtained from the cerebellomedullary cistern (CMC) using a 22-gauge spinal needle (Spinal Needle M type; Hakko, Japan). A total of 2~3 mL of CSF was collected into a plain, sterile tube without anticoagulant. Total nucleated cell count (TNCC) and cytology were performed when there was no blood contamination. Total protein (mg/dL) and albumin (mg/dL) concentrations in the CSF samples collected from the CMC 1 day after ischemic stroke induction along with the albumin quota (AQ) were measured using protein electrophoresis.
2,3,5-Triphenyltetrazolium hydrochloride (TTC) staining and histopathological examination
TTC staining has been used to detect ischemic infarction in tissues [4]. TTC is reduced by certain enzymes in normal tissue to a light-sensitive compound that stains normal tissue deep red. Thus, normal tissue and infarcted abnormal tissue can be clearly distinguished [4].
Except for dogs that died acutely, all canines were euthanized with propofol (6 mg/kg IV, Provive; Myungmoon Pharm) and KCl (0.6 g/kg IV, Kcl-40 inj; Daihan Pharm, Korea) 7 or 14 days after the induction of ischemic stroke. The brains were carefully removed and gross findings were recorded before the brains were cut into 2-mm-thick coronal sections. Fresh brain slices that included the ischemic lesions were immersed in a 2% solution of TTC (Sigma-Aldrich, USA) in normal saline for 30 min at 37℃ as previously described [4,14].
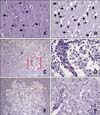
After 2% TTC staining, the sections were immersed in 10% paraformaldehyde with phosphate buffer and fixed for at least 72 h at room temparature. The slices were then dehydrated and embedded in paraffin. Transverse sections (5-µm-thick) were cut and stained with hematoxylin and eosin (H&E) for histopathological examination as follows. The sections were incubated in xylene for 10 min followed by dehydration in different concentrations of alcohol (100%, 100%, 95%, and 70% for 5 min each), rinsed in H2O, placed in Harris hematoxylin solution for 3 min, rinsed in H2O, and then placed in eosin for 2 min (at room temparature). After staining, the sections were washed with a series of different concentrations of alcohol (70%, 90%, 95%, 100%, and 100%, with 10 dips in each), dipped in xylene, and then mounted using permount. At the end of H&E staining procedure, examination for histopathological alterations due to ischemic stroke was performed by light microscopy (BX41; Olympus, Japan).
Immunohistochemical staining for IL-6
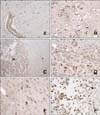
Serial 4-µm-thick brain tissue sections were deparaffinized in xylene, rehydrated in a graded series of ethanol solutions, and washed in phosphate-buffered saline (PBS; pH 7.4; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 2 mM KH2PO4). Endogenous peroxidase activity was blocked by incubation with 3% H2O2 for 20 min at room temperature. Heat-induced antigen retrieval was performed by boiling in Tris-EDTA (pH 9.0) or citric acid (pH 6.0) for 15 min in a microwave. Nonspecific binding was blocked by incubation with a blocking solution (3% fetal bovine serum albumin in PBS). The slides were washed three times in PBS and then incubated with a polyclonal goat antibody against canine IL-6 (1:100; Santa Cruz Biotechnology, USA) to identify the presence and distribution of IL-6. All of the slides were subsequently washed four times in PBS and then incubated with horseradish peroxidase-conjugated secondary antibody (DAKO REALTM Envision kit; Dako, Denmark) for 40 min. Peroxidase activity was visualized using 3,3'-diaminobenzidine tetrahydrochloride (Wako, Japan) and the sections were counterstained with hematoxylin.
Immunohistochemistry images were collected using an Olympus BX41 microscope (Olympus) fitted with a Leica DFC 290 digital camera (Leica, Switzerland). Positive cells were counted under 100× magnification (1.3 mm2) using an image analyzer (Image-Pro Plus; Media Cybernetics, USA). IL-6 expression was quantified by counting the number of cells positive for each antibody in five fields including cerebral cortices.
Enzyme-linked immunosorbent assay (ELISA) for IL-6 in CSF
Each CSF sample was centrifuged immediately after collection at 2000 × g for 10 min in a microcentrifuge (Microcentrifuge 5415R; Eppendorf, Germany). Following centrifugation, the supernatant was stored at - 70℃ (CLN-70UW; Nihon Freezer, Japan) until analysis for IL-6. The IL-6 concentration of each CSF sample was measured using a commercially available quantitative solid-phase sandwich ELISA kit (Quantikine ELISA; R&D Systems, USA) according to the manufacturer's instructions.
Results
Induction of ischemic stroke and assessment of neurological status
Ischemic stroke was successfully induced by permanent MCAO in nine dogs. The animals were divided into three groups: group A (three dogs; survived only 1 day due to severe neurological deterioration), group B (two dogs; euthanized 1 week after the experiment), and group C (four dogs; euthanized 2 weeks after the experiment) according to survival time and/or duration of the experimental period. Most of the dogs showed neurological signs characteristic of forebrain dysfunction, including reduced consciousness (n = 9), head turning (n = 7), circling (n = 6), ataxia with hemiparesis (n = 8), hemianopsia with an absent menace response (n = 9), and nystagmus (n = 1). Neurological scores of the nine dogs were recorded daily (Table 1). Except for nos. 4 and 6, dogs in groups B and C showed gradual improvement over the experimental period without specific therapy.
MRI findings
The first MR images were obtained 1 day after the induction of ischemic stroke. The lateral cerebral cortex (n = 9) and caudate nucleus (n = 8) of the affected side were the main areas affected in most dogs. Involvement of the basal nuclei was observed in only one animal (no. 6). All of the ischemic lesions were well defined, sharply demarcated from adjacent brain parenchyma, and confined to the cerebral grey matter. All lesions showed hypointensity on T1WI and hyperintensity on T2WI, FLAIR, and DWI (Fig. 1). For dogs in groups B and C, serial MR images were obtained according to the predetermined schedule. The volume of ischemic lesions increased between the first (day 1) and second (day 3) MRI scans, and decreased gradually thereafter (Fig. 1). The calculated ischemic lesion volume as a percentage of brain volume on day 1 was significantly higher for group A than groups B and C (p < 0.0238; Table 2). The percent ischemic lesion volume increased between the first (day 1) and second (day 3) MRI scans, and decreased gradually thereafter (Table 2).
CSF analysis
The TNCC (reference, less than 5/µL), cytology, total protein concentration (reference, 10~27 mg/dL), albumin concentration (reference, 5~28 mg/dL), and AQ (reference, less than 0.3) were evaluated. The TNCC was determined and a cytologic examination was performed only when there was no blood contamination. The TNCC of six dogs was evaluated, and all animals had TNCC values above the reference level. Cytologic examination revealed the presence of monocytic pleocytosis in two dogs. CSF total protein and albumin concentrations along with the AQ were measured to estimate the disruption of the BBB in eight dogs. An elevated AQ was observed in all eight canines and there were no significant differences among the groups (0.68 ± 0.28; reference, less than 0.3).
TTC staining and histopathological examination
Gross post-mortem findings were recorded according to the experimental period. Cerebral parenchymal edema and discoloration of the affected side were observed in dogs from group A. In contrast, animals in groups B and C had mild to severe cerebral parenchymal atrophy and necrosis on the affected side without edema. These gross findings were consistent with features observed in the cross-sectional slices. Affected lesions in the cross-sectional slices were not stained with TTC, and the size of unstained lesions were consistent with the MRI findings (Fig. 2).
Results of H&E staining were also recorded according to the experimental period. Variable degrees of cavitation and necrosis due to neuronal cytopathic effects, such as pyknotic nuclei and cytoplasmic shrinkage, were observed on the affected side of the cerebral cortex in all dogs (Fig. 3). Canines in group A exhibited rare inflammatory cell infiltration within the parenchyma or around the blood vessels (panel B in Fig. 3). Macrophage infiltration into the ischemic parenchyma and mononuclear perivascular cuffing were observed in all animals in groups B and C (panels C-F in Fig. 3). Two dogs in group C had marked necrotic lesions with severe vacuolar degeneration relative to the adjacent normal parenchyma (panel E in Fig. 3).
Immunohistochemical staining for IL-6
Immunohistochemical staining was performed to observe the presence and amount of IL-6 within the ischemic lesions. Regardless of the experimental group, all dogs had increased IL-6 immunoreactivity in the ischemic lesions compared to the contralateral unaffected normal parenchyma (Fig. 4). Normal brain parenchyma was negative for IL-6 immunoreactivity. However, neuronal cells of ischemic lesion were positive for IL-6. Dogs in group A frequently exhibited IL-6-positive neuronal cells with pyknotic nuclei and cytoplasmic shrinkage accompanied by cavitation. These findings were also observed in animals from groups B and C in combination with other specific characteristics such as infiltration of IL-6-positive macrophages and mononuclear cells into the parenchyma or around blood vessels (panels C-F in Fig. 4).
IL-6 levels in the CSF measured by an ELISA
The IL-6 concentration in CSF was measured with an ELISA. IL-6 levels at each time point are summarized in Table 3. In particular, the concentration of IL-6 on day 1 correlated positively with the severity of neurological deterioration, including death. The mean level of IL-6 on day 1 was significantly higher for group A than groups B and C (p < 0.0016; Table 3). Changes in the IL-6 concentration over time were also examined (Table 3).
Discussion
In the present study, ischemic stroke was induced by MCAO, and time-related changes of clinical features, MRI findings, CSF IL-6 concentration, histopathology, and tissue expression of IL-6 were evaluated. Ischemic lesion volumes (MRI) and CSF IL-6 concentrations on day 1 were significantly higher in group A compared to groups B and C. Furthermore, expression of IL-6 in ischemic lesions was observed with immunohistochemistry.
We used dogs as a model of ischemic stroke due to their gyrencephalic brains, which are anatomically and functionally similar to human brain. In addition, ease of sampling, relatively large brain size for optimal imaging, and ease of monitoring make dogs an ideal animal model of ischemic stroke [14]. In addition to these advantages, results of the present study had relatively high reproducibility and consistency in terms of lesion induction, lesion size, and clinical signs. However, the present study also had several limitations. The scoring system for neurological signs was too simple to fully accommodate the requirements of clinical evaluation of human ischemic stroke for which diverse scoring systems have been established. Examples of these systems include the National Institutes of Health (NIH) stroke scale, Barthel index, modified Rankin scale (MRS), Canadian neurological scale (CNS), European stroke scale, hemispheric stroke scale, and Scandinavian stroke scale [1,20,36]. These scoring systems designed for humans are far more sophisticated and precise than the one established for dogs. For example, the NIH stroke scale evaluates 11 main categories versus six for the dog scoring system. Another critical limitation of the dog as a model for therapy was the resolution of neurological signs observed in the dogs from groups B and C, which survived the acute ischemic insult.
Because albumin in CSF is exclusively of serum origin, increased CSF albumin levels can be used to evaluate BBB integrity. The amount of albumin in the CSF may vary with serum albumin concentration; therefore, the ratio of albumin levels in CSF to serum albumin concentration, known as the AQ, has been recommended as a more meaningful measure than the CSF albumin concentration alone [23,29]. In a previous study, the AQ of 10 normal dogs ranged from 0.17 to 0.3 [29]. The AQ was measured in eight dogs in the present investigation and was elevated above the normal range (0.41 to 1.06) in all cases. However, similar increases of the AQ have been observed with other CNS diseases accompanied by BBB disruption such as inflammatory, viral, and neoplastic disorders [29]. Therefore, AQ is also insufficient to confirm a diagnosis of ischemic stroke but may be useful for differentiating early cytotoxic edema from later vasogenic edema due to BBB breakdown [27].
Edema formation is a primary cause of poor outcomes and a common cause of death following acute ischemic stroke [27]. The two major types of edema related to ischemic stroke are cytotoxic and vasogenic. Cytotoxic edema occurs immediately after the ischemic episode and is induced by translocation of fluid from the interstitial to intracellular compartment due to ionic and metabolic imbalances. Most of the brain cells consequently swell, but there is no significant increase in the overall brain volume sufficient to cause a mass effect because of the corresponding reduction in extracellular space [27]. In contrast to cytotoxic edema, vasogenic edema occurs mainly because of direct alteration of the BBB integrity. After acute ischemic insult and induction of cytotoxic edema, the BBB is disrupted. This disruption increases BBB permeability so that macromolecules are able to pass through, allowing translocation of fluid from the intravascular compartment to the extracellular space [27]. Therefore, vasogenic edema increases the overall brain volume and tends to be more pronounced in the white matter than in grey matter due to greater compliance of the extracellular compartment of the former [2,27]. Previous human medicine study have demonstrated that the severity of vasogenic edema usually peaks 2 to 5 days after ischemic stroke [27]. In the present study, all dogs in groups B and C, which survived the acute ischemic insult, showed similar increases in volume of lesion between days 1 and 3 according to MRI. Although the animals in group A died 1 day after ischemic stroke, they also had a significantly greater volume of lesion compared to dogs in groups B and C. This means early therapeutic intervention is necessary before vasogenic edema in the infarcted area causes significant volume expansion.
Previous studies have attempted to establish rational time windows for treating ischemic stroke and identifying patients most likely to benefit from early therapeutic intervention by obtaining a detailed understanding of changes in various MR sequences over time [13,18,28,32]. Among these MR sequences, apparent diffusion coefficient (ADC) mapping and diffusion-weighted imaging (DWI) are of particular interest because these methods can detect early ischemic changes in infarcted brain tissue within hours after ischemic insult whereas no significant abnormalities are typically seen on conventional MR sequences such as T2WI, T1WI, and FLAIR. Typical features of acute ischemic lesions have high signal intensity on DWI and low signal levels on ADC mapping [15,36]. Evolution of the ADC over time after ischemia has been studied in animal models [15] and human stroke patients [36]. According to these investigations, ADC values decline rapidly after ischemic insult due to the development of cytotoxic edema and then subsequently increase to normal values as vasogenic edema develops. Changes in DWI over time are less sensitive to acute cytotoxic edema because the latter tends to maintain high signal intensity for a long period [19]. Therefore, a combination of ADC, DWI, and other conventional sequences is recommended to diagnose ischemic stroke, establish a therapeutic plan, and evaluate the overall prognosis. In the current study, ADC mapping could not be used due to the low magnetic field of our MR system and technical problems. Therefore, further studies including advanced MR sequences will be essential to establish a more sophisticated animal model of ischemic stroke.
Examination of H&E-stained tissue sections in the present investigation revealed various histopathological changes associated with ischemic stroke. Findings indicative of acute ischemic stroke, including signs of acute neuronal injury such as pyknotic nuclei with cytoplasmic shrinkage, were commonly observed in all experimental groups. One characteristic histopathological abnormality of groups B and C that was absent in group A was perivascular or parenchymal infiltration of mononuclear cells and macrophages into the brain. These differences were reasonably consistent with previously reported histopathological changes associated with acute ischemic stroke in humans [1,8,20].
According to previous reports [3,8,9,20], histopathological changes associated with acute ischemic stroke can be divided into three major phases: acute neuronal injury (1 to 2 days), organization (3 to 53 days), and resolution (26 days to 23 years). Characteristic histopathologic changes observed during the acute neuronal injury phase are neuronal eosinophilia and other neuronal changes including the formation of pyknotic nuclei and cytoplasmic shrinkage; these were seen in all cases. In our study, acute neuronal changes were observed in all experimental groups after 1~14 days. This persistence of acute neuronal injury may be explained by previous reports showing that acute neuronal changes persist for up to 16 days in squirrel monkeys [9] and up to 35~60 days in humans [8].
Characteristic histopathologic changes found during the organization phase are acute and chronic inflammation within the brain parenchyma and around blood vessels. Acute inflammation, characterized by infiltration of polymorphonuclear leukocytes such as neutrophils, occurs in both experimental animal and natural human stroke, and persists for up to 7 and 37 days, respectively [8,9,20]. In the present study, only dog no. 3 in group A had perivascular infiltration of neutrophils. Approximately 3 days after the onset of cerebral infarction, acute inflammation progressively evolves into chronic inflammation characterized by the presence of mononuclear cells and macrophages, which may persist for up to 53 years [20]. Such chronic inflammatory changes with infiltration of numerous mononuclear cells and macrophages were observed in dogs from groups B and C in our investigation. Taken together, findings from our experimental groups were very similar to those described in previous reports. The changes we documented can be divided into two phases: an acute neuronal injury phase in group A and an organization phase in groups B and C. Further studies examining greater numbers of dogs over longer experimental periods will be needed to better understand the pathogenesis of ischemic stroke in animal models in greater detail.
Since CSF is considered the component most reflective of spontaneous pathologic responses in ischemic brain tissue, most studies of CSF have investigated the pathogenetic role and prognostic value of CSF cytokine levels [25]. In particular, a positive correlation of elevated IL-6 levels in CSF with poor outcomes and ischemic lesion volume has been reported in several studies of human ischemic stroke [33,34]. In the present study, IL-6 concentrations in CSF on day 1 were remarkably higher in group A than in groups B and C. Consistent with higher IL-6 concentrations, dogs in group A had greater lesion volumes and worse outcomes than those in groups B and C. One major question regarding elevated CSF IL-6 levels is whether the IL-6 is produced intracranially by various cells or originates from an extracranial source and enters the brain via peripheral blood through the damaged BBB [33,34]. There are two main hypotheses explaining the source of intracranial IL-6 production [16]. The first is that IL-6 is produced by both damaged neurons and non-neuronal cells after MCAO, which has been observed in rats [31]. The second is that IL-6 is secreted by infiltrating macrophages as well as brain-derived cells such as microglia and astrocytes, for which there is also evidence [22,24]. On the other hand, the extracranial hypothesis requires both a high serum level of IL-6 and disruption of the BBB. In a previous report [33], it was noted that the IL-6 concentrations in stroke patients were significantly lower in serum than CSF. In addition, no significant difference in the CSF IL-6 concentration between patients with intact and disrupted BBBs was found.
In the present study, we monitored disruption of the BBB according to the AQ (normally < 0.3) on day 1, which ranged from 0.41 to 1.06 and did not differ among the experimental groups. However, IL-6 concentrations in CSF on day 1 were significantly different between group 1 and groups B or C. Therefore, the degree of IL-6 elevation in CSF may not be associated with the extent of BBB disruption. Although we did not measure serum IL-6 concentrations in this study, an extracranial source can be excluded based on the AQ values and data from previous reports [33,34]. In addition, immunohistochemical staining revealed extensive IL-6 expression in damaged neuronal cells in the dogs from group A, which also exhibited significantly higher IL-6 concentrations in the CSF. Therefore, findings from our study support the intracranial hypothesis of IL-6 production, especially production by damaged neuronal cells. However, as mentioned above the present study has two major limitations: a relatively small number of subjects and a lack of serum IL-6 data. Further investigations employing quantitative and molecular analyses of a large number of subjects will be needed to elucidate the exact mechanism(s) by which IL-6 concentrations in CSF are elevated during acute ischemic stroke.
In the present study, ischemic stroke was induced by MCAO in 9 dogs and clinical signs, MRI findings, CSF IL-6 concentration, histopathological changes, and tissue expression of IL-6 were evaluated serially. Based on results in this study, CSF IL-6 levels could be the valuable prognostic factor in ischemic stroke patients.




 XML Download
XML Download