

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Actinobacillus (A.) pleuropneumoniae, a Gram-negative bacterium, is the causative agent of necrotizing and hemorrhagic pleuropneumonia in pigs [8,13]. Fifteen serotypes of A. pleuropneumoniae have been identified based on antigenic differences of the capsular polysaccharide [7]. A. pleuropneumoniae exotoxin (Apx) is one of the important virulence factors contributing to the pathogenesis of porcine pleuropneumonia [13]. Given their structural feature, Apx toxins belong to the family of repeats in toxin (RTX) [24]. Other members of the RTX family include hemolysin produced by Escherichia (E.) coli and leukotoxin (LktA) from Mannheimia haemolytica [19,23]. ApxI to IV toxins cause pulmonary damage in pigs [1,15,22]. Among these toxins, ApxI exerts strong hemolytic and cytotoxic effects on a variety of cells including porcine neutrophils, lymphocytes, porcine alveolar macrophages (PAMs), and pulmonary endothelial cells [9,11,14,21,22]. Additionally, ApxI induces apoptosis and pro-inflammatory cytokine expression in PAMs through a mitogen-activated protein kinase pathway [2,3,25].
Our previous study investigated how a single species of exotoxin affects PAMs by using the crude cultural supernatant derived from A. pleuropneumoniae serotype 10 as the source of native ApxI. It was necessary to use either heat-inactivated or antibody-neutralized exotoxin or add lipopolysaccharide (LPS) inhibitor polymyxin B to demonstrate the specificity of apoptotic effect caused by ApxI [2,4,25]. The purpose of the present study was to construct an apxIA mutant using a strain of A. pleuropneumoniae serotype 10 bacterium [10] by inserting a kanamycin-resistant gene cassette into the apxIA gene via allelic exchange. The culture supernatant of this mutant designated ApxIA336 was evaluated for biological activity namely, hemolytic activity; cytotoxicity; apoptotic effects; and virulence to mice.
Materials and Methods
Antibodies and chemicals
Restriction enzymes were purchased from New England Biolabs (USA). Anti-digoxigenin (DIG) Fab fragment conjugated with horseradish peroxidase (HRP), a DIG DNA labeling kit, and cytotoxic detection kit (LDH) were obtained from Roche (Switzerland). Hoechst 33258, nicotinamide adenine dinucleotide (NAD), poly-D-lysine (PDL), polymyxin B, and 2,3-bis (2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide inner salt (XTT) were obtained from Sigma-Aldrich (USA). Bradford reagent and γ-globulin standard were purchased from Bio-Rad Laboratories (USA). A limulus amebocyte lysate (LAL) assay kit was obtained from Cambrex Bio Science (USA). Medium, antibiotics and supplementary components for cell culture were purchased from Gibco (USA). Bacterial culture materials were purchased from Becton Dickinson (USA).
Cell culture
Primary porcine alveolar macrophages (PAMs) were prepared from 4 to 6 weeks old healthy piglets as described elsewhere [25]. The piglets were cared for and euthanized based on a protocol approved by the Institutional Animal Care and Use Committee (IACUC) of National Chung Hsing University (Taiwan, R.O.C.). PAMs were obtained through lavage and stored in liquid nitrogen as previously described [4]. PAMs were thawed and cultured overnight in RPMI 1640 supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin at 37℃ in 5% CO2 before the experiments.
Construction of the apxIA insertion mutant
An apxIA mutant was constructed by introducing a kanamycin-resistant gene cassette into the apxIA gene of A. pleuropneumoniae serotype 10 (strain 13039, Denmark), a kind gift from the Animal Health Research Institute, Council of Agriculture, Republic of China, using a molecular cloning approach (Fig. 1). Briefly, A. pleuropneumoniae serotype 10 was cultured overnight in brain heart infusion (BHI) broth containing 10 µg/mL NAD (NAD/BHI) at 37℃. Genomic DNA was extracted from the bacterial culture according to the manual provided by QIAamp DNA mini kit (QIAGEN, Germany). Full-length apxIA DNA was amplified by PCR using primers ApxI-F1 and ApxI-R1 (Table 1): initial denaturing of DNA at 94℃ for 5 min, followed by 35 cycles of denature at 94℃ for 45 sec, annealing at 53℃ for 45 sec, and extension at 72℃ for 2 min, then a final extension at 72℃ for 7 min. The amplified PCR product was then purified by QIAquick PCR purification kit (QIAGEN, Germany) and ligated to a pCR-XL-TOPO vector (Invitrogen, USA) according to the manufacturer's instructions. The construct was used to transform into One Shot TOP10 E. coli and cultured in Luria-Bertani (LB) broth supplemented with 25 µg/mL of zeocine. pCR-XL-TOPO plasmid containing apxI was referred to as pCRKZApxIA. Next, a suicide vector construct was created by removing the kanamycin-resistant gene of pCRKZApxIA via restriction enzyme digestion with SphI followed by self-ligation of the plasmid. The resulting plasmid DNA was called pCRZApxIA. The pCRZApxIA plasmid was digested with SspI to create a blunt end in the apxIA gene fragment and ligated to a kanamycin-resistant cassette gene from the pUC4K plasmid digested with HincII. The resulting suicide vector used for allelic exchange was named pCRZApxIAK.
Allelic exchange in A. pleuropneumoniae was performed by diluting the overnight culture of A. pleuropneumoniae serotype 10 at a ratio of 1 : 20 in LB broth supplemented with 10 µg/mL NAD and culturing at 37℃ for 3 h with constant agitation. Ten mL of the bacterial culture were then centrifuged at 16,000 × g for 1 min at 4℃, and the resulting pellet was washed three times with and resuspended in 400 µL ice-cold electroporation buffer [27 mM sucrose, 15% glycerol, and 3 mM of potassium phosphate (pH 7.4)] [12]. Five to ten µg of pCRZApxIAK plasmid DNA were added to the bacterial suspension, transferred to a 2-mm electroporation cuvette (Thermo Fisher Scientific, USA), and pulsed at 2.5 kV, 800 Ω, and 25 µF (Bio-Rad Laboratories). Next, 1 mL of NAD/BHI broth was added and the cells were cultured at 37℃ for 3 h. The transformants were further cultured overnight at 37℃ on a NAD/BHI agar plate supplemented with 25 µg/mL kanamycin. Finally, bacterial colonies were analyzed by PCR with different primer pairs (Table 1). DNA amplified with primers ApxI-F1/R1 and kana-F/R was initially denatured at 94℃ for 5 min, followed by 35 cycles of denature at 94℃ for 45 sec, annealing at 53℃ for 45 sec, extension at 72℃ for 2 min, and a final extension at 72℃ for 7 min. DNA amplified with primers ApxIV-F/R was initially denatured at 94℃ for 5 min, followed by 35 cycles of denature at 94℃ for 30 sec, annealing at 62℃ for 30 sec, extension at 72℃ for 3 min, and a final extension at 72℃ for 10 min.
Southern blot analysis
Genomic DNA of parental and mutant strains were extracted and digested with PvuI. Five µg of digested DNA were separated by electrophoresis in 0.7% agarose and transferred onto a nylon membrane (Amersham Biosciences, Sweden). The DNA was crosslinked to the membrane by exposure to ultraviolet light for 2 min. ApxI-F2/R2 primers and DIG-labeled dNTP were then used to generate the apxIA probe by using A. pleuropneumoniae serotype 10 genomic DNA as a PCR template. Next, pUC4K plasmid DNA was amplified using Kana-F/R primers to produce a DIG-labeled kanamycin-resistant cassette probe. The nylon membranes were hybridized overnight with 25 ng/mL of DIG-labeled apxIA or kanamycin-resistant cassette probes at 42℃. The membranes were subsequently washed with buffer containing 100 mM maleic acid, 150 mM NaCl, and 3% Tween 20 (pH 7.5) and probed with an anti-DIG Fab fragment conjugated with horseradish peroxidase at room temperature for 1 h. Probe hybridization was detected using chemiluminescence reagent plus (PerkinElmer, USA).
Sequencing analysis
PCR was performed with genomic DNA from mutant strain using the primer pair ApxI-F1/R1. The PCR product was sequenced by Tri-I Biotech (Taiwan, R.O.C.).
Bacterial growth curve determination and measurement of kanamycin-resistant cassette insertion stability
A single colony of parental or mutant strain was transferred to 10 mL NAD/BHI broth and cultured at 37℃ with constant agitation for 16 h. The culture was mixed with fresh NAD/BHI broth at a 1 : 100 ratio and incubated at 37℃ for 7 h. At 1-h intervals, 100 µL of bacterial suspension was sampled, 10-fold serial-diluted and spread onto NAD/BHI agar plates. Following incubation at 37℃ overnight, the colonies on the plates were counted, and the number of colony forming units (CFUs) per mL was calculated.
Stability of the kanamycin-resistant cassette in mutant strain was also evaluated by growing bacteria in NAD/BHI broth for 10 serial passages. Genomic DNA was extracted from the bacteria at each passage. PCR amplification of the kanamycin-resistant cassette was then performed.
Exotoxin preparation, protein concentration determination, and measurement of lipopolysaccharide (LPS) content
A. pleuropneumoniae exotoxin was prepared from a cultural supernatant as previously described [4]. Protein and LPS concentrations in the exotoxin preparation were measured with a Bradford protein assay and limulus amebocyte lysate (LAL) assay, respectively. Exotoxin derived from the parental strain contained 1.56 mg/mL of protein and 1.02 × 106 endotoxin units (EU)/mL of LPS. Exotoxin from the mutant strain contained 1.50 mg/mL of protein and 9.99 × 105 EU/mL of LPS.
SDS-PAGE and Western blot analysis
Twenty-five µg of total protein from A. pleuropneumoniae exotoxin was separated by SDS-PAGE and stained with Coomassie Brilliant Blue R-250 (Sigma-Aldrich). For Western blot analysis, exotoxin with 2.5 µg protein was subjected to SDS-PAGE and transfer to a polyvinylidene difluoride (PVDF) membrane (PerkinElmer). The proteins were detected with a rabbit antiserum raised against recombinant subunit ApxI [4].
Hemolytic assay
Hemolytic activity of the exotoxin was evaluated according to a protocol described elsewhere with minor modifications [9]. Heparinized whole blood was collected from a pig. Red blood cells (RBCs) were washed with Tris-saline buffer [TS buffer; 10 mM Tris and 0.8% NaCl (pH 7.5)] and resuspended in TS buffer supplemented with 1 mM of CaCl2 (TS/CaCl2) at a ratio of 1 : 100. Exotoxin was 2-fold serially diluted with TS/CaCl2 buffer and incubated with washed RBCs at 37℃ for 2 h. Percentage of hemolysis was calculated by measuring absorbance at 550 nm using a plate reader (Dynex Technologies, USA).
Lactate dehydrogenase (LDH) release and mitochondrial activity assays
Membrane-damaging effects of the exotoxin were evaluated in PAMs using an LDH release assay (Roche, Switzerland) based on the manufacturer's instructions. Briefly, 2.5 × 104 PAMs were incubated with various concentrations of exotoxin at 37℃ for 4 h. Levels of LDH in the culture medium were determined with the LDH assay. Effects of the exotoxin on mitochondrial activity were evaluated in the PAMs with an XTT assay as previously described [4].
Apoptosis scoring
PAMs were plated onto a 12-mm coverslip coated with 25 µg/mL PDL at a density of 2.5 × 105 cells per well and stimulated with bacterial cultural supernatants with protein concentrations of 1, 2.5, or 5 µg/mL at 37℃ for 6 h. Apoptosis was then monitored by evaluating nuclear condensation and/or fragmentation after Hoechst 33258 staining as previously described [4].
Fifty percent lethal dose (LD50) in mice
Six-week-old BALB/c mice were purchased from the National Laboratory Animal Center (Taiwan, R.O.C.). The mice were cared for and handled according to the guidelines of the IACUC of National Chung Hsing University. The mice were intraperitoneally injected with 500 µL of 10-fold serial-diluted bacterial suspension. Mortality was recorded 7 days post-inoculation, and the LD50 was calculated according to the method of Reed and Muench [20].
Statistical analysis
Data were obtained from three independent experiments with at least duplicate determinations. Differences were analyzed using a one-way analysis of variance (ANOVA) with Microsoft Office Excel 2003 (Microsoft, USA). Results are expressed as the mean ± standard error of the mean (SE).
Results
Identification of the apxIA insertion mutant
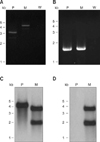
An apxIA insertion mutant was constructed through homologous recombination in A. pleuropneumoniae serotype 10. Genotype of a candidate mutant strain was characterized using PCR, Southern blot analysis, and DNA sequencing. Results of PCR using primers targeting apxIA indicated that the parental strain of A. pleuropneumoniae serotype 10 contained the full-length apxIA gene with a size of 3137 bp. PCR product generated from the mutant strain was 4389 bp, indicating that a kanamycin-resistant cassette had been inserted into the apxIA gene (Fig. 2A). PCR amplification of apxIVA was also performed to confirm that both the mutant and parental strains were A. pleuropneumoniae (Fig. 2B).
Insertion of the kanamycin-resistant gene cassette into apxIA introduced a PvuI restriction enzyme cleavage site within the genomic DNA of the mutant strain (Fig. 1). Southern blot analysis confirmed that only one DNA restriction fragment hybridized with the DIG-labeled apxIA probe in the parental strain while two hybridized restriction fragments were observed for the mutant strain (Fig. 2C). Additionally, a probe specific for the kanamycin-resistant gene hybridized with two DNA fragments from the mutant strain but not the parental strain (Fig. 2D).
Finally, sequencing analysis revealed that the kanamycin-resistant cassette had been inserted into a region immediately after 1005 bp of the apxIA gene in the mutant strain (data not shown). The insertion created a frame-shift mutation in the apxIA gene. The mutant strain was designated as ApxIA336.
Insertion of the kanamycin-resistant cassette does not affect ApxIA336 growth
In this study, we also attempted to determine whether insertion of the kanamycin-resistant cassette into the apxIA gene affected bacterial growth. For this, the mutant and parental strains were cultured, and the growth curves were monitored. Experimental results indicated that the growth rates of both strains were similar and reached a plateau at 5 h post-inoculation (Fig. 3A). Stability of the insertion mutation was further evaluated by identifying the kanamycin-resistant gene by PCR in ApxIA336 cultures subjected to 10 serial passages. A fragment of expected size (633 bp) was observed in bacteria from all 10 passages, and provided evidence of the stability of the insertion in ApxIA336 (Fig. 3B).
ApxIA336 does not produce ApxI
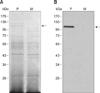
Disruption of ApxI production in the ApxIA336 mutant was demonstrated using SDS-PAGE and Western blotting. SDS-PAGE analysis of bacterial cultural supernatants revealed multiple proteins produced by both the parental and mutant strains (Fig. 4A). Western blot analysis indicated that when applying an antiserum raised against the recombinant subunit ApxI [4], only a single band was observed in the parental strain, but not in the mutant strain (Fig. 4B). This finding confirmed that ApxIA336 failed to produce exotoxin ApxI. Additionally, the levels of protein and LPS in cultural supernatants from the mutant and parental strains appeared to be comparable (see Materials and Methods).
Cultural supernatant from ApxIA336 fails to induce hemolysis
Hemolytic activity of cultural supernatants from both the mutant and parental strains was assessed using porcine RBCs. Notably, ApxIA336 cultural supernatant did not induce hemolysis even at a protein concentration of 200 µg/mL. Conversely, the parental culture supernatant containing 3.1 to 200 µg/mL of protein caused 10 to 100% of the RBCs undergo hemolysis (Fig. 5A). These results indicated that without the production of ApxI, cultural supernatant from ApxIA336 had no lytic effect on porcine erythrocytes.
Cultural supernatant from ApxIA336 does not affect cell membrane integrity or mitochondrial activity in PAMs
Damaging effects of the bacterial cultural supernatants on cell membrane integrity and mitochondrial activity were evaluated. For this, LDH release and XTT assays, respectively, were performed with PAMs as the target cells. Experimental results indicated that cultural supernatant from ApxIA336 did not adversely affect cell membrane integrity or mitochondrial activity even at protein concentrations of 200 µg/mL, the highest concentration tested (Figs. 5B and C).
In contrast, the level of LDH release was elevated with increasing protein concentrations of the parental cultural supernatant (Fig. 5B). At 50 µg/mL, the parental supernatant induced 100% LDH release, indicative of extensive membrane damage to the PAMs. Treatment with the parental supernatant also impaired PAM mitochondrial activity in a concentration-dependent manner. Moreover, parental supernatant containing 12.5 to 200 µg/mL of protein caused a 25 to 100% of decrease in mitochondrial activity (Fig. 5C).
Cultural supernatant of ApxIA336 fails to induce apoptosis in PAMs
The ability of ApxI in cultural supernatant to induce apoptosis was determined by treating PAMs with various concentrations of bacterial cultural supernatants and scoring for apoptosis. Cultural supernatant derived from ApxIA336 with protein concentrations ranging from 1 to 5 µg/mL induced apoptosis at a rate of 3.5 to 4.9%, which did not significantly differ from that of the untreated control group (Fig. 5D). PAMs treated with parental cultural supernatant containing 1, 2.5, or 5 µg/mL of protein induced apoptosis at a rate of 6.2%, 17.1%, and 41.3%, respectively (Fig. 5D). These results indicated that cultural supernatant from ApxIA336 failed to induce apoptosis in PAMs.
The virulence of ApxIA336 is attenuated in mice
Bacterial virulence was evaluated by intraperitoneally injecting BALB/c mice with various concentrations of the mutant or parental strain. The LD50 values of the mutant and parental strains were 2.8 × 108 and 1.9 × 107 CFUs, respectively (Table 2). In the mice, the mutant strain ApxIA336 was 15 times less virulent than the parental strain.
Discussion
Apx exotoxins produced by A. pleuropneumoniae are virulence factors that profoundly impact the pathogenesis of porcine pleuropneumonia [13,21]. Given the heat-labile nature of the exotoxins [13], bioactive native Apx is not easily obtained after purification [5,6]. Our previous study investigated how a single species of exotoxin affects PAMs using crude cultural supernatant derived from A. pleuropneumoniae serotype 10 as the source of native ApxI. Specificity of the apoptotic effect exerted by ApxI has been demonstrated by using several controls, including neutralization with antibody, heat-inactivation of the toxin, and the addition of polymyxin B [2,4,25]. Alternatively, an apxIA-insertion mutant derived from A. pleuropneumoniae serotype 10 would be valuable for further elucidating the effects of ApxI on target cells.
In the present study, ApxIA336 mutant strain was constructed through homologous recombination that resulted in the insertion of a kanamycin-resistant gene cassette into the apxIA gene, leading to the disruption of gene expression. A full-length apxIA gene consists of 3069 bp. The insertion occurred immediately after 1005 bp, which was approximately one-third of the length from the 5'-end of the apxIA gene and caused a frame-shift in the apxIA open reading frame. The disruption of ApxI production in mutant strain was confirmed by Western blot analysis in which rabbit antiserum raised against amino acids 37-626 of ApxI [4] failed to recognize any protein in the ApxIA336 cultural supernatant (Fig. 4B).
The observation that ApxIA336 had a similar growth rate as that of the parental strain indicates that the insertion mutation did not affect overall bacterial growth. This finding is similar to previous data from Xu et al. [26] who constructed a mutant strain of A. pleuropneumoniae serotype 10 by replacing the 1.8-kb gene fragment at the C-terminal of apxIA with a chloramphenicol-resistant cassette. The resulting mutant strain produced a truncated form of ApxI protein, while its growth rate was comparable to that of the parental strain. This same study demonstrated that the mutant strain did not induce hemolysis when cultured on blood agar with Staphylococcus epidermidis. To the best of our knowledge, the cytotoxic effects of exotoxin derived from the mutant strain [26] have not been previously addressed.
The present investigation demonstrated that ApxIA336 did not produce ApxI. However, multiple protein bands were observed in cultural supernatants from both the parental and mutant strains by SDS-PAGE. Additionally, a similar amount of LPS was found in the cultural supernatants of both parental and mutant strains. However, without ApxI, a mixture of multiple proteins and LPS in the cultural supernatant apparently did not cause hemolysis, damage cell membranes, impair mitochondrial activity, or induce the apoptosis of PAMs. These findings support the notion that ApxI, rather than LPS or other components produced by the bacteria, is largely responsible for the hemolytic and cytotoxic effects of crude exotoxin preparations. It is still unclear whether LPS potentiates the effects of ApxI. Nevertheless, a previous study determined that LPS enhances β2 integrin expression on bovine neutrophils, subsequently increasing the cytotoxic effect of M. haemolytica LktA [17].
The ApxIA336 mutant constructed in our study was subjected to virulence analysis in mice. According to the results, virulence of the mutant strain was attenuated compared to the parental strain. The reduction of virulence in mice was similar to the findings from a study by of Xu et al. [26] in which 80% of the mice survived after being challenged with 2 × 108 CFUs of a mutant strain while only 40% of the mice survived a challenge with 2 × 106 CFUs of the parental strain. Notably, in both studies, the parental strain exhibited a higher virulence in mice than the apxIA-mutant strains, indicating a potential role of ApxI in bacterial virulence, despite the fact that mice are not a natural host of A. pleuropneumoniae. Nevertheless, the mouse model has been extensively adopted in evaluating the vaccine efficacy against A. pleuropneumoniae [3,16,18]. Whether the virulence of ApxIA336 is attenuated in pigs warrants further study.
In conclusion, disruption of ApxI production corresponds to the loss of hemolytic and ability to damage cells of the culture supernatant from ApxIA336, indicating a pivotal role of ApxI in these effects. Without ApxI, LPS in conjunction with other components in bacterial cultural supernatant failed to elicit hemolytic or cytotoxic effects towards RBCs or PAMs, respectively. Mutant ApxIA336 is highly promising for use in future studies designed to elucidate how ApxI affects target cells.





 XML Download
XML Download