

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Porcine species have a great impact on studies of biomaterial production, organ transplantation, and specific biomodel development [2,7]. Establishment of porcine parthenogenetic embryonic stem cells (ppESCs) is expected to make significant contributions to therapeutic applications. The advantage of parthenogenetic stem cells is that they can be banked and distributed as "off-the-shelf" embryonic stem cells (ESCs) with less ethical concerns than in vitro or in vivo fertilized oocyte-derived stem cells [9]. While many researchers have attempted to establish immune-matched parthenogenetic ESCs to increase the clinical application of ESCs [28], the low efficiency of producing viable embryos from which to derive the ESCs has made achieving this goal a challenge. In particular, embryo quality decreases after parthenogenetic activation [4,18,22]. Although several culture systems have been suggested for improving the establishment of ESCs [3,25,33,34], previous studies have failed to show sufficient correlation between different culture conditions and improved establishment of ESCs. Therefore, more efficient culture and isolation methods are needed, especially for parthenogenetic embryos that are more sensitive to the culture environment than normally fertilized embryos [20,29]. In the present study, we evaluated the effects of serum-rich and cytokine-supplemented culture conditions on the establishment of ppESCs.
Materials and Methods
Animal care and experiment materials
Ovaries were collected from crossbred (Duroc × Landrace × Yorkshire) prepubertal gilts at a local slaughterhouse and transported to the laboratory in Dulbecco's phosphate-buffered saline (PBS; Invitrogen, USA) supplemented with 1× Pen Strep (penicillin 100 unit/mL streptomycin 100 µg/mL, Gibco-BRL, USA ). All procedures for animal management and surgery followed the standard protocols of Seoul National University (Korea). The Institutional Animal Care and Use Committee Review Board at Seoul National University approved the research proposal (SNU-200908-13). All chemicals were purchased from Sigma Chemical Company (USA) unless otherwise stated.
Oocyte collection and in vitro maturation
Cumulus-oocyte complexes (COCs) were aspirated from antral follicles with a diameter of 3~8 mm using an 18 G needle attached to a 10-mL syringe. Only COCs with multiple layers of compacted cumulus cells were selected. These complexes were then washed three times and transferred to a 4-well dish (ThermoFisher Scientific, USA) containing 500 µL of an in vitro maturation (IVM) medium with gonadotropin including 10 IU/mL pregnant mare serum gonadotropin (PMSG; Intervet, The Netherlands) and 10 IU/mL human chorionic gonadotropin (hCG; Intervet). The cells were incubated at 39℃ with 5% CO2 in a humidified atmosphere [31]. After maturation for 22 h, the COCs were incubated in IVM medium without PMSG or hCG for an additional 22 h.
Parthenogenesis, embryo culture, and blastocyst manipulation
Oocytes that reached the MII stage after 44 h of culturing in IVM medium were activated with two pulses of 120 V/mm DC for 60 µsec in a 280-mM mannitol solution containing 0.01 mM CaCl2·2H2O and 0.05 mM MgCl2·6H2O. Following electrical activation, the oocytes were treated with 5 µg/mL cytochalasin B for 4 h. The activated oocytes were washed three times with fresh in vitro culture medium [IVC medium; porcine zygote medium-3 (PZM-3) supplemented with 0.3% (w/v) fatty acid-free bovine serum albumin (BSA; Bioworld, Korea), 2.77 mM myoinositol, 0.34 mM trisodium citrate (Wako, Japan), and 10 µM β-mercaptoethanol], which was designated as FBS (-) medium. The oocytes were cultured in 30-µL droplets of IVC medium under mineral oil and subsequently cultured at 39℃ in a humidified atmosphere of 5% CO2, 5% O2, and 90% N2 for 7 days. To culture with FBS (Hyclone, USA), the embryos were transferred into freshly prepared IVC medium containing 10% (v/v) heat inactivated FBS (Hyclone), which was designated as FBS (+) medium, on the 3rd day of IVC culturing using a micropipette tip. To artificially eliminate the zona pellucida (ZP), unhatched blastocysts on the 5th and 6th day of IVC culturing were treated with 50 µL of acidic Tyrode's solution (0.24 mg/mL CaCl2·2H2O, 0.1 mg/mL MgCl2·6H2O, 0.2 mg/mL KCl, 8 mg/mL NaCl, 1 mg/mL d-glucose, and 4 mg/mL polyvinylpyrolidone; pH 2.5 ± 0.3) for 5 min.
ICM implantation onto feeder cells and ppESC culturing
ZP-free blastocysts obtained by both spontaneous and artificial hatching processes were washed three times. The ICMs were then separated from the trophectoderm by gently pipetting with fine-pulled glass capillary pipettes in embryonic stem cell (ES)-basal media [1:1 mixture of low-glucose Dulbecco's Modified Eagle Medium (DMEM) (Invitrogen) and Ham's F-10 medium (Invitrogen), 2 mM l-glutamine, 1% (v/v) non-essential amino acids (Invitrogen), 0.1 mM β-mercaptoethanol (Invitrogen), and 1% (v/v) penicillin-streptomycin (Invitrogen)] containing 15% (v/v) FBS (HyClone) supplemented with 20 ng/mL recombinant human basic fibroblast growth factor (bFGF; Invitrogen), 20 ng/mL recombinant human stem cell factor (SCF; R&D Systems, USA), and 20 ng/mL recombinant human leukemia inhibitory factor (LIF) before use. From this point, the cells were cultured at 37℃ with 5% CO2 in a humidified atmosphere.
Isolated ICMs were implanted on a monolayer of mitomycin C-treated mouse embryonic fibroblasts (MEFs) that were isolated using general methods [21]. After implantation, the culture medium was refreshed every other day. Attachment efficiencies of the ICMs were measured on the second day after implantation. Eight to 12 days after the first implantation, ICM-derived colony-forming cells (ppESCs) were mechanically removed from the MEFs and reseeded onto new MEFs. Next, the ppESCs were sub-cultured every 4 days and the culture medium was changed every day.
Alkaline phosphatase (AP) activity assay
Cells were fixed with 4% formaldehyde for 30 min and washed with PBS. After incubation with staining solution provided in a BCIP/NBT Alkaline Phosphatase Substrate Detection Kit IV (Vector Lab, USA) for 1 h at room temperature, the cells were washed with PBS and examined under an Eclipse Ti-U inverted microscope (Nikon, Japan).
Immunofluorescence (IF) staining
ICM-derived colony-forming cells (ppESCs) were sub-cultured into new MEFs on glass cover slips (Marlenfeld, Germany) in 24-well culture plates (TPP, USA). After 4 days, the cells were fixed with 4% formaldehyde for 20 min at room temperature, permeabilized with 0.4% Triton X-100 in PBS for 30 min, and washed with PBS-T buffer (PBS containing 0.05% Tween-20). After incubating with blocking buffer (PBS-T buffer containing 3% BSA) for 1 h at room temperature, the cells were incubated overnight at 4℃ with each primary antibody diluted 1:100 in blocking buffer. Species reactivity of the primary antibodies had been verified in previous reports and the following primary antibodies were used: rabbit anti-octamer-binding transcription factor 3/4 (OCT3/4; Santa Cruz, USA) [19], goat anti-Nanog (Abcam, UK), mouse anti-stage-specific embryonic antigens (SSEA4; Millipore), mouse anti-cytokeratin-17 (CK17; Millipore) [8,24], mouse anti-vimentin (Millipore), mouse anti-desmin (Millipore) [3], mouse anti-cytokeratin 8-18-19 (CK8-18-19; Abcam), mouse anti-α smooth muscle actin (SMA; Abcam), and mouse anti-microtubule-associated protein 2 (MAP2; Millipore). The cells were washed three times with PBS-T buffer and then incubated with the following secondary antibodies diluted 1:100 in blocking buffer for 1 h at room temperature: Alexa fluor 488-conjugated goat anti-rabbit IgG (Invitrogen), rhodamine-conjugated rabbit anti-goat IgG (Invitrogen), Alexa fluor 647-conjugated goat anti-mouse IgG (Invitrogen), or Oregon green 514-conjugated goat anti-mouse IgG (Invitrogen). After washing three times with PBS-T buffer, mounting solution with Hoechst 33258 (Invitrogen) was used for nuclear staining. Negative controls were produced by incubation with secondary antibody alone. The stained cells were observed under a Carl Zeiss Axiovert 200M inverted fluorescence microscope (Carl Zeiss, Germany).
Reverse transcription (RT)-PCR
Total RNA was extracted from the cells using an RNeasy Mini Kit (Qiagen, Germany). cDNA synthesis was performed with an RT kit (Promega, USA). DNA amplification was conducted with forward and reverse primers specific for the following factors using a Go-script PCR kit (Promega): porcine OCT3/4 (forward): 5'-AG GTGTTCAGCCAAACGACC-3', (reverse): 5'-TGATCG TTTGCCCTTCTGGC-3', porcine Nanog (forward): 5'-ATCCAGCTTGTCCCCAAAG-3', (reverse): 5'-ATTTC ATTCGCTGGTTCTGG-3', porcine GATA-binding factor-6 (GATA-6; forward): 5'-CAGGAAACGAAAACCTAAGA GCAT-3', (reverse): 5'-TTCTCGGGATTAGCGCTCTC-3', porcine neurofilament-H (NF-H; forward): 5'-AGAGCT GGAGGCACTGAAAA-3', (reverse): 5'-TCCGACACT CTTCACCTTCC-3', porcine bone morphogenetic protein-4 (BMP-4; forward): 5'-TGAGCCTTTCCAGCAAGTTT-3', (reverse): 5'-CAACGCACAGATCAGGAAGA-3', porcine glyceraldehyde 3-phosphate dehydrogenase (GAPDH; forward): 5'-TCGGAGTGAACGGATTTG-3', (reverse): 5'-CCTGGAAGATGGTGATGG-3'. PCR was performed at 94℃ for 5 min for initial denaturation followed by 27 cycles of 60℃ for 40 sec and 72℃ for 1 min before a final extension at 72℃ for 5 min. GAPDH was used as a reference gene. The amplified products were analyzed on 2% agarose gels and product size was confirmed with a DNA ladder.
Telomerase activity assessment
Telomerase activity of undifferentiated or differentiated ppESCs (ppESC1 and ppESC2) was measured using a TeloTAGGG PCR ELISA kit (Roche, USA) according to the manufacturer's protocol. The result was analyzed with positive control (Human embryonic kidney [HEK] 293 cells) and negative control (heat treated HEK 293 cells, lysis reagent and synthetic oligonucleotide).
Measurement of embryoid body (EB) formation and further differentiation
ppESCs were detached from the MEFs and further dissociated by incubation with 0.125% trypsin/EDTA for 5 min at 37℃. For EB formation, the single cells (approximately 3,000 cells/EB) were transferred to AggreWell400 plates (Stem Cell Technologies, USA) and then cultured for 4 days in ES-basal medium containing 10% (v/v) FBS (Hyclone) without bFGF, SCF, or LIF. Next, the EBs were transferred to culture dishes coated with 0.1% gelatin for further differentiation, and maintained in ES-basal medium including 10% (v/v) FBS without bFGF, SCF, or LIF that was refreshed every other day. After 4~5 days of culturing, differentiated cells were used for RT-PCR analysis and IF staining.
Teratoma formation and histological analysis
ppESCs were treated with a 10% (v/v) accutase solution (Invitrogen) in PBS (Invitrogen) for 2 min at 37℃. The cells were the separated by gently pipetting up and down five to six times. Dissociated ppESCs were neutralized after cell counting with three different solutions: PBS alone, ES-basal media containing 15% (v/v) FBS (Hyclone), and ES-basal media containing 15% (v/v) FBS (Hyclone) with bFGF, LIF, and SCF. For transplantation, 6-week-old immune-deficient NOD-SCID mice (Charles River, Japan) were maintained under non-specific pathogen-free conditions. The animals were anesthetized with 2% (v/v) isoflurane. Next, 100 µL of the ppESC solution containing 5 × 106 cells combined with same volume of pre-chilled Matrigel without phenol red (Invitrogen) was directly injected into the subcutaneous region of the recipient mice using a 28.5-gauge insulin syringe. The mice were sacrificed after tumours larger than 1 cm3 had developed. The tumours were collected, and tumour invasion into other organs including the liver, lungs, and spleen was evaluated. For histological analysis, teratoma tissues were fixed with a 10% (v/v) formaldehyde solution for 2 days and then stained with hematoxylin and eosin (H&E). Expression of markers for the three germ layers was monitored with IF staining.
Results
Effects of FBS on blastocyst development
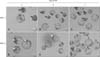
Blastocysts that were cultured in the IVC medium alone were poorly developed. Many had irregular blastomeres and fragments in the perivitelline area (panels A-C in Fig. 1). Conversely, blastocysts maintained in the FBS (+) medium were well developed compared to those grown in the IVC medium alone during the same period (panels D-F in Fig. 1). ICMs were positioned appropriately in each group of blastocysts (panel D in Fig. 1). On the 6th day of IVC, blastocysts in the FBS (+) medium were more mature and had begun to hatch spontaneously from the ZP (panel E in Fig. 1). On the 7th day of IVC, most blastocysts in the FBS (+) medium had matured into fully developed blastocysts and were completely hatched (panel F in Fig. 1).
Effects of FBS treatment on ICM attachment rates
Attachment rates of ICMs isolated from the FBS (+) group were higher than those of the FBS (-) group (Table 1, 40% vs. 24%). Since we observed a tendency for the FBS (+) group to spontaneously hatch relative to the FBS (-) group (data not shown), we compared the attachment efficiency of spontaneously hatched and artificially hatched blastocysts within the FBS (+) group. ICMs derived from spontaneously hatched blastocysts had a higher attachment rate than those derived from artificially hatched blastocysts (Table 2, 42% vs. 25%) and generated primitive ppESC colonies (Table 2, 39% vs. 0%). Hence, we decided to use the spontaneously hatched blastocysts cultured in FBS (+) medium for further experiments.
Effects of cytokines on ICM attachment rates
We evaluated the effect of cytokines on ICM attachment efficiency and propagation of ICM-derived cells. For this, the cells were cultured under four different conditions (Table 3). The attachment efficiency of ICMs in bFGF- and LIF-supplemented medium (Medium II) was greater than that of ICMs maintained with bFGF alone (Medium I; 56% vs. 11%) or bFGF and SCF (Medium III; 56% vs. 34%). The best ICM attachment rate (82%) and greatest formation of primitive ppESC colonies were observed with the bFGF, LIF, and SCF combination (Medium IV; Table 3). Therefore, we selected Medium IV as the culture medium for the first ICM implantation and further culturing.
Characterization of ppESC stemness
Seven ppESC lines were established from spontaneously hatched blastocysts from the FBS (+) group and maintained in Medium IV. Although the trophectoderm cells remained around the ICMs transferred onto MEFs (panel A in Fig. 2), most disappeared between 8 and 12 days after implantation. The ICM-derived colony-forming cells were flat and round (panel B in Fig. 2). After a second sub-passage, the colonies had clarified margins (panel C in Fig. 2). AP activity was detected in all ppESC lines but no reactivity was detected for MEFs around the ppESCs (panel A in Fig. 3). Additionally, all cell lines were positive for stemness markers (OCT3/4 and Nanog) by RT-PCR and IF staining (panels B and C in Fig. 3). Expression of the stem cell surface marker SSEA4 was also detected (panel C in Fig. 3). All of these proteins were highly expressed. Our ppESCs also had high levels of telomerase activity (panel D in Fig. 3) that disappeared after differentiation. The normal porcine chromosome number, 38XY (n = 38), was found in our ppESCs (unpublished data).
Characterization of the ppESC differentiation capacity
Our ppESCs were able to form EBs (panel A in Fig. 4). mRNA expression of markers for the three germ layers (i.e., NF-H, BMP-4, and GATA6) was highly increased in the EBs. On the other hand, expression of stemness markers (OCT3/4 and Nanog) was dramatically decreased (panel B in Fig. 4). The protein expression of lineage-specific cytoskeleton components (vimentin, desmin, and cytokeratin-17) was also detected in each of the differentiated cells (panel C in Fig. 4).
Among all the NOD-SCID mice subcutaneously injected with ppESCs, only group C (ppESCs in the injection medium with bFGF, SCF, and LIF) produced teratomas (Table 4). Typical structures representing the three germ layers, perineurium of the involved nerve, blood vessels, and several tubular epithelial structures formed within the granulation tissue layer (panel A in Fig. 5) were observed in all ppESC-derived teratoma tissues (panel A in Fig. 5). The expression of lineage-specific markers (CK8-18-19, SMA, and MAP-2) was also confirmed (panel B in Fig. 5).
Discussion
ICMs that give rise to definitive fetal structures are generated during the early developmental stage of blastocysts, and ESCs originate from these cell masses [32]. ICM attachment onto feeder cells is the first important step for the production of ESCs from embryos [23]. Therefore, many researchers have explored various culture conditions to derive ICM and obtain the ESCs [30]. However, it is difficult to culture and maintain ICMs in vitro.
In previous reports, FBS was shown to exert inhibitory effects on the early cleavage of embryos, but promote the rapid development of morulae into blastocysts, yielding higher quality blastocysts [6,17]. Moreover, the ability of blastocysts to transition into the 4-cell stage and subsequent hatching was dependent on the length of time for which the cells were cultured in FBS-free IVC medium before transfer to FBS-supplemented medium [6,12]. In addition, when fetuses produced in FBS free-medium or medium containing BSA and medium containing FBS from morulae were transplanted into the uterus, successful implantation and further development were achieved for a higher proportion of fetuses produced from blastocysts cultured in FBS-containing medium [12]. Consistent with these previous reports, we also observed the positive effect of FBS on ICM attachment and ESC generation.
Several chemicals and hormones such as insulin are present in FBS [26]. In previous studies of ESC establishment, insulin was found to improve the development and viability of embryos [13,15]. In addition, insulin increases the ICM cell number of blastocysts [14] and persistence of outgrowth of the implanted cells to generate ESCs [5]. Therefore, it was speculated that although we did not check the ICM cell number or hatching rate, insulin in the FBS administered during blastocyst development may have enhanced embryo viability and blastocyst development, resulting in high ppESC attachment and production rates in our study.
Recently, there have been many reports about the effects of cytokines on embryo development and ESC maintenance. FGF is needed for maintenance of human ES cells [1]. SCF is known to have inhibitory effects on apoptosis that are induced by treatment with Fas-L, and enhancing effects on cell recovery during embryo development in vitro [10,11]. Consistent with these findings, we observed more viable embryos in SCF-supplemented media (data not shown). In addition, enhancement of cell attachment for blastocysts and bovine embryonic stem-like cells during later passages was observed when culturing with bFGF, SCF, and LIF [16]. Likewise, we observed the same effects on attachment of ICMs and pluripotency maintenance of long-term ppESC cultures up to passage 52.
In our study, we used Matrigel to produce teratomas in mice. Matrigel that consists of extracellular matrix proteins and growth factors has been known to enhance teratoma formation by elevating graft survival and growth rates due to endovascularization to the injection site as well as interaction between the extracellular matrix of the recipient and the injected cells [27]. However, teratoma formation in this study was not significantly affected by the Matrigel. Instead, the addition of bFGF, SCF, and LIF to the Matrigel-based injection medium significantly improved the teratoma formation efficiency. Although further experiments are needed to explain these effects, a more suitable environment that promotes the survival of xenogeneic cells might be achieved by co-injection of LIF, bFGF, and SCF.
In summary, we demonstrated the usefulness of FBS treatment during blastocyst development and ICM attachment. We also observed the effects of LIF, bFGF, and SCF with ES-basal medium on enhancing the attachment of ICMs as well as ppESC generation and long-term culturing. This study is the first to report teratoma formation from ppESCs delivered by injection with the three cytokines. Our data suggest that improved efficiency for ppESC generation using our technique will contribute to the investigation of porcine stem cell biology, and is a safe and reliable method for producing porcine ESCs for clinical applications.








 XML Download
XML Download