

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Hepatocellular carcinoma (HCC) is a primary malignancy of hepatocytes which accounts for 80% of all primary liver cancers and ranks globally as the fourth leading cause of cancer-related death [1]. HCC is a major public health issue in Korea due to its high incidence related to chronic infection with hepatitis B and C virus [19]. In addition to viral infections, several etiological risk factors have been identified including alcohol abuse, expose to aflatoxin B1, and nonalcoholic fatty liver change [16]. HCC frequently arises in patients with liver cirrhosis, the result of a complex pathogenesis involving chronic hepatitis, necrosis, and regenerative fibrosis. Since terminal stage HCC can rarely be cured by conventional therapeutic approaches, the final remaining therapeutic choices are surgical resection and transplantation [13]. However, the survival rate of HCC patients who have undergone surgical resection is known to be less than 25% [31]. Therefore, development of an effective cancer chemopreventive agent is urgently needed for hepatic cancer prevention. Cancer chemoprevention is defined as an attempt to suppress or prevent the carcinogenic progression with natural or synthetic chemical agents. These agents might prevent or reverse the carcinogenic progression during the premalignant or early stages by inducing the apoptosis of cancerous cells or inducing differentiation.
The elm tree (Ulmus davidiana var. japonika Nakai) is a deciduous tree that is naturally widespread throughout Korea, Japan, and China. The bark of the stem and root of this plant have long been used in Oriental medicine to treat inflammation, edema, mastitis, cancer, and rheumatoid arthritis [22]. The phytochemical components of bark extracts of elm tree (BEE) include (+)-catechin, catechin rhamnoside, catechin apiofuranoside, triterpene esters, sesquiterpene o-naphthaquinones (mansonones E, F, H), lignan, and neolignan glycosides [15,27]. Methanolic extracts of the elm tree exhibit strong anti-oxidant activity in animal models including 1,1-diphenyl-2-picrylhydrazyl radical scavenging and hydroxyl radical scavenging [12], and inhibit endogenous nitric oxide synthesis in macrophages [11]. Moreover, the plant glycoprotein (approximately 116 kDa) has biologically active functions that include anti-oxidative properties, the ability to enhance immune activity [20]. This compound also inhibits the activation of inducible nitric oxide synthase, cyclooxygenase-2, and matrix metalloproteinase-9 by blocking activator protein-1 and nuclear factor-kappa B in lipopolysaccharide-stimulated HCT-116 cells [23]. Mansonone E and F, two sesquiterpene O-naphthoquinone compounds isolated from BEE, exert anti-proliferative and pro-apoptotic effects on HeLa cells that involve the down-regulation of Bcl-2 and Bcl-xL along with the up-regulation of Bax, thereby leading to the initiation of the mitochondrial apoptotic pathway [30]. Additionally, mansonone E was found to have anti-tumor activities in a nude mouse xenograft model of colon cancer and in uterine cancer cells [21]. The anti-cancer properties of BEE were also observed in cancer cell lines and a 1,2-dimethylhydrazine-induced rat colorectal carcinogenesis model [5,18]. In one of the latter studies, BEE decreased the incidence of aberrant crypt foci and reduced crypt multiplicity, thus inducing apoptosis in the region of the colon where most of the pre-neoplastic lesions had accumulated [18].
Although the attributes mentioned above are germane to the use of elm tree as a traditional medication, the mechanism(s) of BEE action should be thoroughly elucidated before applying this compound to treat human health problems. In particular, no study of the modulating effects of BEE on the growth of hepatic cancer cells has been documented. The present study was therefore performed to investigate the effects of BEE on the growth of HepG2 human HCC cells. The effects of BEE on apoptosis and possible impact on the apoptotic pathway were studied.
Materials and Methods
Isolation of BEE chloroform faction
An elm tree was purchased from the Eoleumgol Agricultural Cooperative Union, Korea. A voucher specimen was deposited at College of Agriculture, Life and Environment Sciences, Chungbuk National University, Korea. Fresh root bark (10 kg) was dried in a dark and well-ventilated place. The air-dried root bark samples were milled and underwent an extraction process three times with 80% ethanol at 25℃ at 3.78 × g under reflux. The ethanol extract of elm tree root was filtered through Whatman filter paper (No. 2; Maidstone, England), and concentrated in a rotary vacuum evaporator (N-11; Eyela, Japan) at 40℃. The yield of the ethanol extract was 1.41% of the raw material mass. For solvent fractionation, BEE (4 g) was suspended in water, and then underwent a series of extractions successively with equal volumes of n-hexane, chloroform, ethyl acetate, and n-butanol, leaving a residual aqueous fraction. Each fraction was evaporated in vacuo to yield the residues of the hexane (15.7%), chloroform (27.3%), ethyl acetate (7.2%), butanol (29.6%), aqueous (20.2%) fractions. The final components in chloroform-soluble fraction of the ethanol extract were used for the experiment.
Cell culturing
HepG2 human HCC cells were obtained from the Korean Cell Line Bank (Korea). The cells were cultured in Dulbecco's modified Eagles's medium (Hyclone, USA) supplemented with 10% fetal bovine serum (Hyclone, USA), 100 U/mL penicillin, and 100 µg/mL streptomycin (GIBCO, USA) at 37℃ in a 5% CO2 humidified atmosphere.
Cell viability assay
HepG2 cells were plated at a density of 1 × 104 cells/well in 96-well plates (BD Biosciences, USA). Each well contained 100 µL of culture medium. To determine the appropriate concentration that was not cytotoxic, cells cultured for 24 h were evaluated using a cell counting kit-8 (CCK-8) assay kit (Dojindo Molecular Technologies, USA) according to the manufacturer's instructions. Briefly, 10 µL of a CCK-8 solution was added to each well. The plates were incubated for 3~4 h at 37℃. The resulting color was analyzed using a microplate absorbance reader (BioTek, USA). Each assay was carried out in triplicate. The optical density (OD) was measured at 450 nm and the percent viability was calculated as (OD of the drug-treated sample/OD of the non-treated sample) × 100.
4,6-diamino-2-phenylindole (DAPI) staining
Apoptotic cells were identified by DAPI staining. After overnight culturing in an 8-well plate (BD Biosciences, USA) (4 × 105 cells/mL), the HepG2 cells were treated with various concentrations (0, 50, 100 and 200 µg/mL) of BEE in fresh culture medium at 37℃ for 24 h. The cells were then fixed with 500 µL fixing solution (acetic acid : methanol; 1 : 3) for 5 min, dried, and stained with the DNA-specific fluorochrome DAPI (2 µg/mL; Sigma-Aldrich, USA). Following 10 min of incubation, the cells were washed with phosphate buffered saline (PBS), air-dried, mounted with 90% (v/v) glycerol, and examined using a fluorescence microscope (Olympus, Japan).
Quantification of apoptotic cells by flow cytometry
BEE-induced apoptosis in HepG2 cells was determined after labeling with fluorescein isothiocyanate (FITC) annexin-V stain supplied in an apoptosis detection kit (BD Biosciences, USA) that was used according to the manufacturer's instructions. The labeled cells were analyzed using flow cytometry. Briefly, after overnight culturing in a 6-well plate (1 × 105 cells/well; BD Biosciences, USA), HepG2 cells were treated with 0, 50, 100, or 200 µg/mL of BEE in fresh culture medium at 37℃ for 24 h. Both floating and adherent cells were concentrated and suspended in binding buffer (10 mM HEPES, 140 mM NaCl, and 2.5 mM CaCl2, pH 7.4). The mixture was then incubated for 20 min in the dark at room temperature and analyzed by fluorescence-activated cell sorting (FACS) using a FACSCalibur instrument (BD Biosciences, USA).
Western blot analysis
Cultured cells were washed twice with 1 × PBS followed by the addition of 1 mL of PBS. The cells were then scraped into a cold 1.5 mL microtube. Cells were homogenized in 100 µL lysis buffer (50 mM Tris pH 8.0, 150 mM NaCl, 0.02% sodium azide, 0.2% sodium dodecyl sulfate (SDS), 1 mM phenyl methane sulfonyl fluoride, 10 µL/mL aprotinin, 1% IGEPAL 630, 10 mM NaF, 0.5 mM EDTA, 0.1 mM EGTA, and 0.5% sodium deoxycholate; Sigma-Aldrich, USA) and centrifuged at 23,000 × g for 15 min. The protein concentration was measured using the Bradford method (Bio-Rad Laboratories, USA) and equal amounts of proteins (40 µg) were separated by 10% or 12.5% SDS-polyacrylamide gel electrophoresis. The resolved proteins were transferred to an immobilon polyvinylidene fluoride membrane (Millipore, USA). The membranes were blocked for 1.5 h at room temperature with 5% (w/v) non-fat dried milk in a Tris-buffered saline (10 mM Tris, pH 8.0 and 150 mM NaCl) solution containing 0.05% Tween-20. The membrane was incubated for 2~5 h at room temperature or overnight at 4℃ with antibodies against inducible Bax, Bcl-2, poly (ADP-ribose) polymerase (PARP), caspase-3, and caspase-9 (1 : 500~1,000 dilution; Cell Signaling Technology, USA). The blot was then incubated with the approperiate horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG (1 : 2,000~3,000 dilutions; Cell Signaling Technology, USA). Immunoreactive proteins were detected with the West-zol Western Blot Detection System (iNtRON Biotechnology, Korea). The relative density of the protein bands was measured by densitometry using Image J (National Institutes of Health, USA).
Results
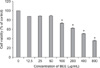
BEE suppresses the viability of HepG2 cells
HepG2 cells were treated with 0~800 µg/mL of BEE for up to 24 h after which cell viability was analyzed with a CCK-8 assay. Starting at a concentration of 100 µg/mL, BEE significantly suppressed the viability of the HepG2 cells in a dose-dependent manner (p < 0.05; Fig. 1).
BEE induces apoptotic cell death
To ascertain whether the BEE inhibition of cell growth was due to the induction of apoptosis, changes in chromatin morphology of the HepG2 cells were examined. HepG2 cells were exposed to 0, 50, 100, and 200 µg/mL of BEE, and DNA fragmentation was detected by DAPI staining. After incubating for 24 h, enhanced DNA fragmentation and apoptotic bodies were observed in cells treated with a high dose (100 and 200 µg/mL) of BEE (Fig. 2). To quantify the number of apoptotic cells treated with BEE, a FITC annexin-V flow cytometry analysis was performed. A representative flow cytometric analysis showed that the percentage of apoptotic cells were 4.48%, 12.10%, 23.52%, and 36.45% of the HepG2 population treated with 0, 50, 100, and 200 µg/mL BEE, respectively (Fig. 3A). BEE significantly increased the number of apoptotic cell deaths in a dose-dependent manner (p < 0.05; Fig. 3B).
Expression of apoptosis-related proteins
Since Bax and Bcl-2 play a crucial role in the apoptotic pathway, we examined the effect of BEE treatment on the expression of these two proteins. BEE treatment enhanced the expression of the pro-apoptotic protein Bax while the level of the anti-apoptotic protein Bcl-2 decreased in a dose-dependent manner (Fig. 4). Since the alteration of Bax/Bcl-2 is known to initiate caspase-9 signaling, the effect of BEE on caspase-9 and caspase-3 was evaluated. BEE treatment resulted in increased expression of cleaved caspase-9 and caspase-3 in a dose-dependent manner although a significant difference was observed only for caspase-3 expression (Fig. 4). Additionally, cleavage of PARP (116 kDa), one of the substrates of activated caspase-3, was measured. Increased levels of an 85-kDa PARP fragment was observed a dose-dependent fashion after incubating for 24 h.
Discussion
Chemoprevention is defined as the use of natural or synthetic compounds to prevent or suppress the progression of cancers. Numerous efforts are being devoted worldwide to discover natural compounds that have potential to suppress cancer cell growth. Many herbal products are available as traditional medicines for cancer prevention. However, not much is known about their efficacy, active principles, mode of action, side effects, or possible adverse interactions with conventional antitumor agents. Therefore, in vitro testing of herbal agents in cancer cell lines may be helpful for understanding their mechanism of action and discovering potential effects. Two different mechanisms are usually employed in cancer chemoprevention: modulation of cell proliferation and induction of cell differentiation.
The present study examined the chemopreventive effects of ethanolic elm tree extract on a hepatic cancer cell line. In order to screen for cytotoxic or apoptotic activities, this study initially determined whether BEE is capable of inhibiting cell proliferation or inducing apoptosis. The colorimetric CCK-8 assay showed that the numbers of formazan-producing cells decreased with increasing doses of BEE; 50% and 66% cytotoxicity was observed with 400 and 800 µg/mL of BEE, respectively. In addition, morphological changes typical of apoptosis were observed in BEE-treated cells. The presence of apoptotic bodies defined as fragments of condensed nuclei was confirmed by DAPI staining. The number of apoptotic bodies increased in a dose-dependent manner. Morphological changes in cell shape and chromatin condensation are the basic and classical criteria for identifying apoptotic cells [14]. These changes are caused by the activation of caspases, proteolytic enzymes that are central factors of apoptosis [4].
Apoptosis is a mechanism in all metazoans for regulating tissue homeostasis through the elimination of redundant or potentially deleterious cells. The induction of apoptosis is arguably the most potent defense system against cancer. For example, immune cells use apoptosis to destroy cancerous cells [10,29], and most chemotherapeutic agents inhibit tumor cell proliferation by inducing apoptosis [3,26]. The triggering of apoptosis, referred to as the initiation phase, is highly dependent on cell type and apoptotic stimuli such as receptor-ligand binding, oxidative stress, radiation, and cytokines [17]. In the subsequent effector phase, cells undergo distinct biochemical changes that result in the systematic activation of procaspase-8, -9, and other factors [7,25]. Activation of these enzymes leads to the execution of related to apoptotic factors and degradation phase through the cleavage of proteins such as caspase-3 and fragmentation of DNA [8,29]. Most of the recent advances in elucidating the apoptotic pathways have come about through the characterization of effector mechanisms. Effector mechanisms of apoptosis have several components. Two effector mechanisms associated with caspase activation have been extensively characterized: the extrinsic or death receptor-mediated mechanism, and the intrinsic or mitochondrial-mediated mechanism [9]. In addition to the mitochondria, other organelles including the endoplasmic reticulum, golgi apparatus, and lysosomes may also have a role in sensing cell damage, pro-apoptotic signaling, and caspase activation [6]. The extrinsic apoptotic pathway is activated at the cell surface when a specific ligand binds to its corresponding cell surface death receptor.
The intrinsic apoptotic pathway relies solely on the permeabilization of mitochondrial membranes to release apoptogenic mitochondrial proteins such as cytochrome c [24] and apoptosis-inducing factor (AIF) [28]. Cytochrome c released into the cytosol triggers the caspase-dependent apoptotic pathway [24]. In contrast, AIF induces caspase-independent apoptosis [33]. In this pathway, Bcl-2 family members appear to play an important role in the regulation of apoptosis. With cell stress, anti-apoptotic Bcl-2 family members (e.g., Bcl-2 and Bcl-xL) residing in the outer mitochondrial membrane can be destabilized by the induction of pro-apoptotic bcl-2 family members (e.g., Bax, Bad, and Bak). In this scenario, the increased ratio of pro-apoptotic Bcl-2 family members to anti-apoptotic Bcl-2 family members induces the release of cytochrome c which binds to Apaf-1, leading to activation of procaspase-9 and subsequent activation of procaspase-3 [3,28,29]. Activated caspase-3 is the key mediator of apoptosis that induces the cleavage and inactivation of key cellular proteins such as PARP [29,32].
In the present study, flow cytometry and DAPI staining showed that BEE treatment induced a significant accumulation of apoptotic cells and increased the number of apoptotic bodies in HepG2 cells. BEE (200 µg/mL) significantly increased the ratio of Bax/Bcl-2 protein in HepG2 cells as detected by Western blot analysis, suggesting that BEE induces apoptosis via the intrinsic mitochondrial-dependent pathway. However, the increase of activated caspase-9 and PARP was not significant although activated caspase-3 levels significantly increased. These findings indicate that BEE possibly induced apoptosis via the caspase-9-independent pathway as well. The exact mechanism of apoptosis induced by BEE should be further elucidated. In particular, it will be important to determine whether and by how much the expression of molecules involved in the caspase-9-independent pathway, such as AIF, is affected.
Regarding the active compounds included in BEE, several candidate compounds such as bakuchiol, mansonone E and F, glycoproteins, and catechins were documented [2,20,27,30]. Those compounds are known to have diverse biological effects including anti-inflammatory, proapoptotic, and anti-oxidative properties according to the isolation method or type of cell line [20,30]. In particular, bakuchiol and masonone E and F are known to be major components in ethanolic BEE that exert anti-inflammatory or pro-apoptotic effects [2]. Therefore, we assume that these compounds might have been included in the BEE and influenced our results.
In summary, we found that BEE inhibited the growth of HepG2 cells by activating the mitochondrial pathway of apoptosis. These results suggest that elm tree root extract may have therapeutic potential for suppressing liver carcinogenesis. The various components of BEE responsible for these actions should be determined by future studies.



 XML Download
XML Download