

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Within the Bacillus genus, the most pathogenic species is Bacillus (B.) anthracis, the causative agent of anthrax and a Gram-positive spore-forming soil bacterium [9,28]. Anthrax is a disease that occurs worldwide. This zoonosis and infectious disease affects a wide range of species, including humans and herbivorous mammals [3,32]. The incidence of anthrax has been reduced by continuous research and disease control, but still occurs in undeveloped and developing countries.
In 1905, the first anthrax case occurred in animal was recorded in Korea. During the next 5 years after the first official report in 1907, the number of anthrax cases occurred in animal increased to 2,562, causing serious damage to related industries that handle animal by-products like meat and animal skins. More than 1,000 cases were subsequently reported annually, and until 1924 the rate of occurrence was approximately 500 cases per year [22]. The reduction of anthrax cases is due to development of live vaccines using non-virulent B. anthracis Sterne and other methods of disease control. However, there were two cases reported in 1994 (Kyungju and Hongseong, respectively), one in 1995 (Hongseong), another two cases in 2000 (Changnyung), and one in 2008 (Yeongcheon). The disease occurs annually in a seasonal manner, but the distribution of cases is most prominent in May and August. Since the annual average rainfall is high in April and July to August, it can be inferred that contaminated soil is more exposed during the rainy season and pollutes pastures, thus transmitting the pathogen to cattle [23].
B. anthracis strains are genetically monomorphic with low levels of sequence diversity [18,26]. Discriminatory markers have long been used to identify and subdivide groups of these microbial pathogens. Tracking the epidemic and pandemic spread of particular strains may be possible using such markers, yielding a long-term overview of epidemiological patterns [1]. There are regions in the B. anthracis genome that vary greatly from strain to strain. Numerous studies have shown that genetic markers such as single-nucleotide polymorphisms (SNPs), variable number tandem repeats (VNTRs), and single-nucleotide repeats (SNRs) can be used to discriminate between strains in B. anthracis [2,5,7,12,15].
Canonical SNPs (canSNPs), originally described by Keim et al. [12], are a number of SNPs located at key phylogenetic junctions and are sufficient to create phylogenetic trees. It is evident that canSNPs can identify relationships among global B. anthracis isolates. Previously, a set of canSNPs was able to distinguish three major lineages of B. anthracis isolates [36] and 12 clonal sub-lineages.
In the present study, we examined a genetic population of B. anthracis isolates from Korea and compared it to a global genetic population using an eight-loci multiple-locus variable-number tandem repeat analysis (MLVA) and 13-canSNP analysis.
Materials and Methods
The B. anthracis strains used in this study included four obtained from the Korean Centers for Disease Control and Prevention (KCDC), five reference strains, and 17 isolates from soil of four different regions in Korea (Table 1). ATCC14185 and ATCC14578 stain obtained from American Type Culture Collection (ATCC), (USA), and Pasteur, delta Sterne, and Sterne strain obtained from National Veterinary Research and Quarantine Service (NVRQS), (Korea). The B. anthracis strains were cultured in brain heart infusion media (Becton Dickinson, USA) and grown at 37℃ with shaking at 200 rpm for 3 days. Total genomic DNA was extracted was modified by Hunter's method [8]. To isolate total bacterial DNA, bacteria cells were harvested by centrifugation at 15,000 rpm for 10 min at 4℃ and resuspended in 1 mL TE buffer (pH 7.4) containing lysozyme (0.1 mg/mL), SDS and proteinase K at 55℃ for 3 h. After the completion of this incubation, DNA was extracted with phenol-chloroform, precipitated with isopropanol and dissolved in TE buffer with RNase (20 µg/mL).
In order to determine whether B. anthracis isolates were full virulent (pXO1+ and/or pXO2+), we performed a polymerase chain reaction (PCR) assay with a primer set specific for B. anthracis [4]. Each PCR mixture (20 µL) contained 10 ng of genomic DNA, 10 pmole of each B. anthracis specific primer, and 2× EF-Taq Premix II (10 µL, SolGent, Korea). The following thermal cycling (Bio-Rad, USA) conditions were used: an initial denaturation step (94℃ for 5 min) followed by 30 cycles of denaturation at 94℃ for 1 min, annealing at 40℃ for 1 min, and primer extension at 72℃ for 1 min. The PCR reaction concluded with a final incubation at 72℃ for 5 min. The PCR products were electrophoretically separated on 1% agarose gel and stained with ethidium bromide.
MLVA was performed for the eight different VNTR loci described by Keim et al. [14]. After amplification, the PCR products were diluted 1 : 10 (volume : volume) in TE buffer and 1 µL of the dilution was added to a mixture containing 8 µL of Hi-DiTM formamide (Applied Biosystems, USA) and ROX-labeled MapMaker 1000 size standard [BioVentures, USA; 20 : 1 (volume : volume)]. The DNA was denatured at 95℃ for 5 min and then immediately placed on ice. Fragments were detected using an ABI 3100 Prism Genetic Analyzer (Applied Biosystems, USA) with a 36-cm capillary at a running voltage of 15 kV at 60℃. To determine the size of the detected fragments, peak height and peak area was measured using GeneScan Analysis software (ver. 3.7; Applied Biosystems, USA). For genotype determination, an MLVA bank developed by the University of Orsay (University Paris XI, France) was used.
For canSNP analysis, we used the primers described by Van Ert et al. [37] and purchased from Genotech (Korea) in which each primer was added a T7 primer sequence as a 5' tail on the primers for sequencing purposes. After amplification, the PCR products were cloned using a T-A cloning vector (RBC Bioscience, Taiwan). Each PCR mixtures contained 2× EF-Taq Premix II (Solgent, Korea), forward and reverse primers (0.5 pmol each), and genomic DNA (400 ng/µL). The thermal cycling conditions were incubation at 95℃ for 5 min that was followed by 30 cycles of 95℃ for 1 min, 60℃ for 1 min, and 72℃ for 1 min. The plasmids were then purified with Hi-Yield Plasmid Mini Kit (RBC Bioscience, Taiwan) and sequenced using the T7 primer. Sequencing analysis was performed five times. MLVA and canSNP analysis data obtained for B. anthracis isolates from Korea were used to calculate simple matching coefficients using MEGA (ver. 4.0.2).
Results
To investigate the genetic diversity of B. anthracis strains isolated in Korea, we used MLVA and canSNP analysis to characterize 21 strains, including four that were obtained from the KCDC (Table 1). Many researchers reported the Pasteur strain to be pXO1- and pXO2+, but the one used in our study was pXO1- and pXO2- since pXO2 was cured during the culturing process. B. anthracis strains obtained from the KCDC were pXO1- and pXO2+ while the isolates obtained in Korea (HS, Bch, KJ, and CH) contained both pathogenic plasmids (pXO1+ and pXO2+; Table 1).
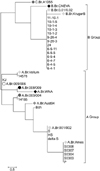
Genotype analysis was performed using eight-locus MLVA data acquired from an MLVA bank. B. anthracis strains obtained from the KCDC were placed into the A3b cluster while the CH isolates were in a B1 cluster (Fig. 1 and Table 2). On the other hand, HS, Bch, and KJ strain were classified as A3a cluster and were not a part of the A3b or B1 clusters. B. anthracis strains obtained from the KCDC (S0303, S0304, S0307, and S0308) had the same MLVA type with #07 strain of Ryu's study. Interestingly, all of the local isolates (CH) were pathogenic and contained identical sequences at eight hypervariable MLVA loci. Sequence repeats in eight of the VNTR loci (vrrA, vrrB1, vrrB2, vrrC1, vrrC2, CG3, pXO1, and pXO2) were identical to those of the other Korean B. anthracis strains from Changnyung (#02 strain of Ryu' study). In our analysis, sequences of the B. anthracis strains obtained from the KCDC (pXO1-, pXO2+) were identical in the VNTR loci (vrrA, vrrB1, vrrB2, vrrC1, vrrC2, and CG3) and varied at one VNTR locus (pXO2) with respect to the Sterne strain. Furthermore, the genotypes of the B. anthracis strains obtained from the KCDC and the Sterne strain differed and were varied at three VNTR loci (vrrA, vrrB1, and vrrB2) with respect to the ATCC14185 and ATCC14578 strains (Table 2). The HS strain had the same MLVA type as #50 strain of Keim's study except vrrC1. The MLVA type of the Bch strain was identical to #58 strain of Keim's study except pXO2. Additionally, the MLVA type of the KJ strain was same as #32 strain of Keim's study except vrrC1.
canSNPs were used to calculate simple matching coefficients and subdivided the B. anthracis isolates according to major sub-lineages (A and B). These were further divided into six of 12 different sub-lineages (Table 3 and Fig. 2, diamond) or sub-groups (circle). In order to make comparisons with global genetic populations, data obtained for B. anthracis Korean isolates from our study along with the results of an eight-loci MLVA and 13-canSNP analysis for 1,033 reported B. anthracis isolates from 42 countries were used to calculate simple matching coefficients (Figs. 2 and 3).
Discussion
B. anthracis VNTR sequences found in many different chromosomal and plasmid locations exhibit a few or even dozens of different allelic states, showing that this bacterium has a degree of diversity frequently lacking among pathogenic microbes [16]. MLVA is currently utilized to identify different sub-populations and particular strains of interest [7,13,14]. For B. anthracis, six chromosomal loci and one locus on each of pXO1 and pXO2 have been used for this technique [14]. In the study of Keim et al. [15], a global B. anthracis (over 400 strains) analyzed and subdivided two major clonal lineages (A and B) and 86 unique MLVA 8 genotype using eight-loci MLVA. This eight-loci MLVA has also been used to examine the genotypes of B. anthracis in France [5], Poland [6], Italy [4], and countries in southern [14] and northern Africa [24]. In 2007, Van Ert et al. [36], used a 15 marker-loci, MLVA15, to examine a collection of 1,033 B. anthracis isolates from 42 countries. This group described three major lineages (A, B, and C) that were further subdivided into 12 clonal sub-lineages or sub-groups, and finally 211 unique MLVA15 genotypes. Le Flèche et al. [21] augmented the MLVA8 assay by proposing 14 additional markers. Additionally, Lista et al. [24] expanded the MLVA21 assay with the addition of four markers (25-loci MLVA) and described two new branches, D and E.
The eight-locus MLVA was used in our study of vrrB1, vrrB2, vrrC1, vrrC2, and CG3 identified by sequencing B. anthracis amplified fragment length polymorphism markers [12]. vrrA was identified by Andersen et al. [2], and the remaining two loci were identified by analysis of the pXO1 and pXO2 plasmid sequences [27]. vrrA and vrrB are located in genes encoding hypothetical protein. vrrC is located in a gene encoding a Ftsk-SpooIIIE DNA translocase family homologue. vrrA, vrrB, and vrrC markers are found in protein coding regions of the B. anthracis genome. As the MLVA marker expands or contracts, the amino acid composition of the protein will be altered [11,31]. This genotyping method had been previously used for consistent subspecies typing worldwide for discrimination of the B. anthracis genome with the highly monomorphic nature [13,14,24]. Among the isolates analyzed in our study, eight VNTR markers had different levels of variability in four loci (vrrA, vrrB1, vrrB2, and pXO2) and were much more variable than other loci.
Based on the MLVA profiles, Korean B. anthracis isolates were assigned to both the A and B branches. Similar genetic dissimilarity has been found in B. anthracis isolates originating from several geographic regions in Africa, Europe, North America, and South America [14]. In a previous analysis, B. anthracis isolates originating from Asia, Turkey, and China were assigned to branch A [36]. It is interesting that our Korean isolates belong to both the A and B branches although our data agree with the results of a previous study [30].
Variations in the number of repeat sequences at a given locus, or in-sequence heterogeneity among individual isolates, allows discrimination among strains. These may be due to slipped-strand mispairing and can occur in combination with inadequate DNA mismatch repair during replication [35]. However, the discriminative power of VNTR markers becomes limited when analyzing strains of close geographical origins [25]. In such situations, diversity can be demonstrated by either analyzing a greater number of VNTR markers [24] or using more sensitive markers [34,36].
SNPs have been used in several studies as important markers that linked sequence variations to phenotypic changes, thus elucidating the molecular basis of diseases [2,7,11,19]. These are also considered extremely valuable to perform phylogenetic analysis as SNPs have very low mutation rates and are less prone to homoplasy [15]. Comparative full-genome sequencing between eight strains of B. anthracis which led to the discovery of about 3,500 SNPs was reported in the study of Read et al. [29] along with 990 SNPs in 26 diverse isolates [15]. These results demonstrate the usefulness of a few strategically placed SNPs that may replace a large number of SNPs for typing B. anthracis strains and generating an SNP-based tree.
canSNP have also been previously used to successfully differentiate B. anthracis isolates and identify worldwide patterns of distribution in which the major clonal lineage A had been shown to be widespread globally [36]. In the study of Van Ert et al. [36], B. anthracis isolates are subdivided into two previously recognized distinct major sub-lineages (A and B) according to the canSNP data, and further divided into 6 of 12 different sub-lineages. In this study, data from an eight-locus MLVA also clearly classified these strains into two major lineages. The more common genotype among the B. anthracis strains previously analyzed and globally distributed strains is the A genotype [36]. However, location of the distinct sub-groups is geographically restricted. Strains obtained from the KCDC (S0303, S0304, S0307, and S0308) and Pasteur strain belong to the A.Br.Ames sub-group, which is found in central and eastern China [30]. The Ames strain was obtained in Texas (USA) in 1981. Later, other isolates were shown to be closely related to the Ames strain [17], indicating the rarity of this strain in nature. Despite its scarcity, the Ames strain is widely used as a reference strain in many laboratories for genotyping analysis.
Two similar strains of B. anthracis (14578) from the KCDC and from the laboratory stock used in our study were found to belong to the A.Br.Vollum sub-lineage. This sub-lineage genotype is dominant in southern Africa and is also present in Europe [33,36]. Three strains (Sterne, HS, and delta Sterne) were found to belong to the A.Br.001/002 sub-group, and a similar genotype has been identified in central and eastern China [36]. canSNP analysis of one laboratory strain (14185) showed that it belongs to the A.Br.003/004 sub-group in which is common in South America.
One of the B. anthracis isolates (KJ) collected recently from Korea belonged to the A.Br.005/006 sub-group, which is common in southern Africa [36]. The Korean B. anthracis isolates from CH analyzed in our study belonged to the B.Br.001/002 canSNP sub-group. This sub-group is primarily found in southern Africa [14]. The major clonal lineages found in Asia are usually assigned to lineage A, which is more common than the other lineages and distributed globally. In contrast, distribution of B lineage isolates is more geographically restricted. B lineage isolates have been found in southern Africa (B.Br.Kruger sub-lineage and B.Br.001/002 canSNP sub-group genotypes), some parts of Europe (B.CNEVA-9006 sub-lineage), and a small region in California, USA [5,6,15]. Additionally, a major clonal lineage B genotype was identified in Korean isolates from CH in this study, and these isolates were closely related to the B1 cluster of MLVA reported by Ryu et al. [30].
Anthrax is rare among humans but very common among grazing animals due to the natural spore transmission cycle. Spores that are formed in a place where an animal died from anthrax are the main source of infection for grazing animals. It is also plausible that livestock grazing over dry, dusty, and contaminated soil inhale spores, leading to infection. Humans can be infected through contact with diseased animals or animal products depending on the route of spore entry: cutaneous (the most frequent form of natural infection), inhalation, and gastrointestinal. Throughout history, anthrax had been widely spread around the world through the trade of infected live animals or meat, hides, hair, wool, or bones from such animals. Infected animals and products are often transported in long distances for industrial, food, or handicraft purposes, and the anthrax spores may find their way to livestock in the local community, thus spreading the disease in locations distant from the original infection source [3]. Restricting the geographical distribution of B. anthracis is primarily a result of niche specialization associated with adaptive differences among the different lineages [10,14]. However, genetic evidence indicates that human activities have dramatically influenced the global population structure of B. anthracis [14,20,36]. Given that natural outbreaks of anthrax are extremely rare, the introduction of new lineages in Korea might be the result of agricultural development and increased international trade. Results of the present study have added further insight into B. anthracis phylogeny and the distribution of Korean isolates.





 XML Download
XML Download