

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Genetically modified (GM) animals are unique mutants used to study gene function and regulation, human and animal diseases, and other biological mechanisms. Cryopreservation of pre-implantation embryos or spermatozoa is a common method for preserving and handling the increasing number of GM mouse lines, often consisting only of very small populations. GM lines must be protected against unexpected disasters. Only after a successful quality assessment of cryopreserved samples has been performed a line can be removed from a breeding nucleus. Both cryopreservation and innovative assessment strategies result in a significant reduction of the number of laboratory animals used as postulated by the 3R principles (replacement, reduction and refinement of animal experiments) as published by Russell and Burch [20]. Since shipping cryopreserved samples is much easier than the transport of living animals, cryotechniques are becoming increasingly important.
For mice, cryopreservation of pre-implantation embryos was historically more successful than cryopreservation of spermatozoa [23,25]. In recent years, methods for cryopreserving spermatozoa have been improved [17]. Both approaches have specific advantages and disadvantages, and the technique used must be selected on a case-by-case basis. However, in vitro fertilization (IVF) required for recovering a line from frozen spermatozoa is a complex procedure [27], and success is often influenced by external factors such as genetic background, the transgene expressed by the line, osmotic stress, the culture media used, and environmental conditions. [4,11,24,28].
Since several parallel sperm samples can be obtained from a single male donor, one or more of these samples can be used for monitoring purposes [29]. Many assays that can determine sperm quality are only motion-based similar to tests performed in reproductive medicine. In the case of mice, the amount of material available for quality assessment is restricted to a few µL. Consequently, commercial kits for evaluating spermatozoa of large animals or humans are not suitable for small laboratory rodents. State-of-the-art assessment of cryopreserved spermatozoa from small laboratory rodents involves IVF followed by embryo transfer [17,23,26]. Both of these procedures are sensitive and can be negatively influenced by the environment [5,14,15]. In addition, they require extensive resources, especially for assessment purposes. To replace these common resource-consuming methods, the goal of this work is the investigation of alternative techniques.
Viability, concentration, motility, and morphology of spermatozoa in a specimen are characteristics that need to be monitored to ensure spermatozoa quality [19]. Motility and concentration can be determined microscopically whereas viability can be monitored with suitable dyes using additive (dyes that stain either viable or dead cells) or subtractive (dyes staining all cells including viable and dead cells) strategies. The staining properties (staining of all, viable, or dead cells) of a dye depend on membrane integrity of the cell or nucleus as well as the ability of the dye to permeate those membranes [28].
In general, a sample can be stained by different dyes, including fluorescent ones. When staining a sample with two or more fluorescent dyes, one has to take care that the emission frequencies of the dyes used will not interfere with one another. Fluorescence-based techniques are very sensitive and allow detection of weak signals, but are also susceptible to background artifacts. We therefore conducted the present study to develop a fluorescent microscopy technique to monitor spermatozoa quality. This strategy could replace the traditional IVF and embryo transfer methodologies. Reliable parameters were identified to create a simple protocol for assessing the quality of (frozen/thawed) spermatozoa.
Materials and Methods
Animal care and housing
All mice used in this study were housed in the animal facility of the German Cancer Research Center (Germany). GM mice lines originally received from different sources were bred and expanded in-house whereas wild-type (WT) mice with corresponding genetic backgrounds (BDF, C3H, C57BL/6, CBA, DBA/2, FVB/N, NMRI) were received from Charles River (Germany). 149 sperm donors, 745 oocyte donors, and 112 foster mothers were included in this study.
Individually ventilated caging systems (IVC) and specified pathogen-free (SPF) facilities (barrier with open caging systems) were used as previously described in detail [5]. Ages of the male mice used ranged between 3 and 9 months. Males were housed alone and females were kept in groups of five. Health of the animals was monitored according to the Federation of European Laboratory Animal Science Associations (FELASA) recommendations [16]. Most mice were housed in an IVC facility that was maintained under SPF conditions. During this study no infectious agents listed in the FELASA guidelines were detected. Animal experimentation was performed according to the German Animal Welfare Act and the Cornell Center for Animal Resources and Education. All animal experiments were approved by the Animal Welfare Department of the Competent Authority (Regierungspräsidium Karlsruhe, Germany) and conducted under the surveillance of the intramural Animal Welfare Committee of the German Cancer Research Center.
Spermatozoa were cryopreserved according to method of Ostermeier et al. [17]. In brief, 3 to 9 months old males were sacrificed by cervical dislocation. Spermatozoa were then collected from the epididmides and vasa deferentia. Spermatozoa were allowed to disperse from the tissue for 10 min at 37℃ in cryoprotective media (CPM) [18% (w/v) raffinose (Sigma-Aldrich, USA), 3% (w/v) skim milk (BD Diagnostics, USA), and 477 µM monothioglycerol (Sigma-Aldrich, USA) in distilled water] and were loaded into 0.30 mL French straws (IMV Technologies, France). The straws were sealed with an impulse tong sealer (Polystar 110 GE/150 D; Rische + Herfurth, Germany), placed onto a polystyrene raft floating in liquid nitrogen (LN2) for at least 10 min, and then stored in LN2. All GM donor males were subjected to re-genotyping by the working-groups responsible for the individual lines.
Sample storage
Cryopreserved samples were stored in the liquid phase of LN2. Storage of the straws was performed according to Schwab and Schenkel [22].
Spermatozoa staining
For staining, spermatozoa were thawed and pre-incubated for 1 h at 37℃. Spermatozoa were stained with two dyes: Hoechst 33342 stain for all spermatozoa (0.001 mg/mL; Invitrogen, USA) and propidium iodide (PI) (M3181.0010, 0.0001 mg/mL; Genaxxon, Germany) to identify cells with compromised membranes. Both dyes we used can stain spermatozoa of several species [3,13,30,33]. A Hoechst 33342 stock solution (10 mg/mL) was stored at -20℃ in the dark. A PI stock solution (1 mg/mL) was stored at 4℃ in the dark no longer than 6 months. Both dyes were dissolved in distilled water. In another, not successful approach, spermatozoa with intact membranes were stained with fluorescein diacetate (FDA) (0.001 mg/mL; Sigma-Aldrich, USA) and spermatozoa with compromised membranes were stained with ethidium bromide (EtBr) (0.001 mg/mL; Applichem, Germany).
A suspension of spermatozoa (20 µL) and 1 µL of each dye were carefully mixed in a 1.5-mL amber microcentrifuge tube. This solution was incubated at 37℃ for 5 min. Next, 10 µL of the suspension were spotted onto thoroughly cleaned and pre-warmed slides (R. Langenbrinck, Germany). Fluorescence microscopy was carried out with an inverted microscope with appropriate filters (wavelength of the emitted light; Hoechst 33342: 461 nm, PI: 617 nm) by using a fluorescence microscope (Axio Observer Z1; Carl Zeiss AG, Germany).
Fluorescence microscopy
The slide containing the spermatozoa suspension was covered with a pre-warmed 18 × 18 mm2 coverslip and placed onto an inverted microscope (TissueFAXSi, Tissue Gnostics, Austria). This technique covered reproducibly the whole volume, no solution or cells were lost, and air-bubbles never appeared under the coverslip. To evaluate motility, a video sequence was taken using Pixelink Capture OEM software and a Pixelink 622 camera (Pixelink, Canada). Dark field illumination was used to acquire 200 frames (1 msec each) at ×5 magnifications. For the membrane integrity assay, 25 fields of view were automatically acquired (with a ×40 objective lens) with TissueFAXSi software controlling the camera (PCO270xs, PCO, Germany), filters, and automatic stage movement (Märzhäuser, Germany). Typical exposure times were 110 ± 30 msec (DAPI filter) and 270 ± 20 msec (Texas Red filter). The entire process including fine adjustment of camera settings did not take longer than 5 to 7 min, and all measurements were taken for the same sample.
Analysis of fluorescence microscopy data
Fluorescence microscopy images were processed with TissueQuest software (TissueGnostics, Austria). First, all spermatozoa were identified by Hoechst 33342 stain. Cell density was expressed in fluorescence-activated cell sorting-like scattergrams where each dot represented one cell. On the y-axis Hoechst 33342 mean staining intensity of every cell was plotted vs. the identified cell size on the x axis (Hoechst 33342 stained area). In this scattergram spermatozoa were identified according to their size, doublets and small debris were excluded from further analysis. All positively identified spermatozoa were then displayed in a second scattergram as Hoechst 33342 mean intensity vs. PI mean intensity. Cut-off values for distinguishing PI-positive from PI-negative cells were set according to the negative controls. Images of cells with PI-staining intensities near the cut-off value (as shown in the scattergram) were evaluated visually to verify and fine-tune the position of the cut-off for each sample individually if necessary. Cells stained exclusively by Hoechst 33342 (those with intact plasma membranes) fell under the cut-off line while double-stained cells (ones without an intact plasma membrane) fell above the cut-off line. As images with known size were analyzed, the resulting quadrant statistics of the second scattergram showed density of spermatozoa (events/mm2), total number of spermatozoa and percentage of living/dead spermatozoa.
To calculate the concentration of spermatozoa in cells/µL we used the values from the scattergrams and calculated the volume as following: 10 µL of the homogenous spermatozoa solution were spotted onto a slide and covered with an 18 × 18 mm2 coverslip. Taking the volume of the staining solution and the dilution factor into account, 1 mm2 corresponded to 0.28 µL or 3.56 mm2 represented 1 µL. This procedure was confirmed by counting spermatozoa in a Neubauer improved counting chamber (Carl Roth, Germany).
In vitro fertilization
IVF was performed as previously described by Ostermeier et al. [17]. Donor females (3~4 weeks old; 5 donors/IVF) with a genetic background according to the individual sperm donor (see Tables 1 and 2) were superovulated as previously described [14,15,22]. These animals were sacrificed 12 to 14 h after the administration of human chorionic gonadotropin (50 IU/mL; Sigma-Aldrich, USA) and the oocytes were removed from the swollen ampullae. Human tubal fluid (HTF) was used as IVF culture medium (Millipore MR-070-D; EMD Millipore, USA). The medium-drop was overlaid with mineral oil (Sigma-Aldrich, USA) as described in [22]. The IVF-dishes (Becton Dickinson, USA) contained one 500-µL HTF drop. Sperm samples were thawed in at 37℃ (in a water bath) for about 10 min. The spermatozoa in CPM were pushed out of the French straw into the HTF drop and were incubated for 1 h at 37℃. Co-incubation of oocytes and spermatozoa in HTF was lasting 5~6 h, the resulting zygotes were washed and incubated overnight in a 200-µL drop of potassium simplex optimization medium (KSOM) (Millipore MR-020P-5F; EMD Millipore, USA). Next, the proportion of two-cell embryos was calculated. These embryos were subsequently used for embryo transfer, cryopreservation, or continued culturing in KSOM.
The success of an IVF was determined by counting two-cell embryos out of all oocytes used for this IVF. However, the two-cell embryos were afterwards used for embryo-transfer, in vitro culture or cryopreservation.
Statistical analysis
Correlations between different parameters were calculated using SAS software (ver. 9.2; SAS, USA) [6]. Differences between two groups of data were analyzed by Spearman's rank-sum test [12]. p-values < 0.05 were considered significant while p-values < 0.001 were highly significant. The influence of membrane integrity (i.e., % of PI-negative spermatozoa in a sample) on fertility (IVF success = % two-cell embryos among all oocytes used for IVF or expanded for the successful recovery of a mouse line) was measured with cross-classified tables using Fisher's exact test [7] with the prerequisite that at least 30 embryos sufficient for two embryo transfers were generated. The coefficient of variation (CV) was defined as the ratio of standard deviation to the mean. CV < 1 indicated low variance while CV > 1 corresponded to high variance [10].
Results
The purpose of the current study was to develop an alternative technique for the common IVF procedure to monitor the quality of cryopreserved mouse spermatozoa. To identify parameters that significantly impact this procedure, various analyses were performed. We evaluated spermatozoa from both WT and GM mice.
Staining results
To determine the number of viable spermatozoa in a sample, we first used an additive strategy involving staining with FDA (specific for viable cells) and EtBr (for dead cells). However, this approach did not produce satisfactory results since spermatozoa do not have enough cytoplasm for these types of staining and develop auto-fluorescence at the excitation wavelength suitable for FDA (450~490 nm, data not shown). Consequently, we chose a subtractive approach in which spermatozoa were treated with two dyes: one that stained all cells (Hoechst 33342, Fig. 1A) and another specific for cells with a compromised membrane (PI, Fig. 1B). This allowed us to determine the number of viable and dead spermatozoa.
Acquisition and analyses of fluorescence microscopy data
Twenty-five fields of view were examined in our fluorescence microscopy analysis. Signals from both dyes in each field of view were analyzed. Merged images were used to identify spermatozoa with intact (blue) or compromised membranes (simultaneous blue and red staining, Fig. 1C). To exclude false-positive signals (e.g., doublets, small debris, or due to contamination) only signals of appropriate size were included into these analyses, the cut-off values for discriminating PI-negative from PI-positive spermatozoa were verified manually in each case (Fig. 2A). Scattergrams allowed us to calculate the amount of PI-negative spermatozoa [% of lower right in Fig. 2B]. As described above, the scattergram allowed us to calculate the concentration of spermatozoa, too. To measure sperm motility, 200 frames (1 msec) were acquired as a video sequence. All video sequences were taken at the same level (Fig. 3). For analyses, the total number of cells and the number of moving spermatozoa were counted, and the ratio of moving spermatozoa was calculated.
Fluorescence microscopy and IVF outcomes
The parameters of spermatozoa determined microscopically were compared with the outcome of IVF by using the same sample. Concentration, motility, and membrane integrity data were correlated with two-cell-embryos received from IVF by Spearman's range-sum correlation. Fig. 4A shows the correlation between spermatozoa concentration and IVF outcomes (p = 0.587). Fig. 4B illustrates the correlation between spermatozoa motility and IVF outcomes (p = 0.5733), and Fig. 4C shows the correlation between PI-negative spermatozoa and the IVF results (p = 0.0001). Our results demonstrated that only membrane integrity was significantly correlated with the outcome of IVF.
The production of two-cell embryos was considered as successful IVF outcome. These embryos were afterwards used for additional purposes. Following over night culture of 108 randomly selected IVF samples, 89.91% of two-cell embryos developed to the eight-cell stage without significant two-cell arrests. Others were cryopreserved or transferred into pseudo-pregnant foster mothers to assess the capacity to recover a GM line in a qualitative approach. GM litters resulting from embryo transfer were identified by genotyping (data not shown).
Reproducibility of the role assessment strategy
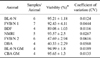
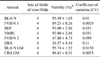
As noted above, positive IVF outcomes were most dependent upon membrane integrity of the spermatozoa. Membrane integrity was further evaluated to determine whether this factor is a reliable marker for sperm viability. The quality of frozen/thawed samples might vary from mouse to mouse. To verify that one out of several samples from the same donor mouse can reliably represent all samples from this donor, 40 samples prepared from eight donor males were analyzed. As demonstrated in detail in Table 1, different samples of the same donor were prepared, spotted onto different cover-slides and were analyzed microscopically. Only a low variance (CV < 1) was detected in all cases. To omit the consequences of a possible in-homogeneity of the material within a slide, four different sets of fields of views (25 fields of view/set) on the same slide were examined. As shown in Table 2, data for eight mice (out of these two different GM lines) with different genetic backgrounds were proven to be homogeneous (CV < 1).
Discussion
The quality of cryopreserved specimens must be assessed [29]. In the present study, we developed a fluorescence microscopy-based technique that can potentially replace the current IVF assays and subsequent embryo transfer to determine the quality of frozen/thawed GM mouse spermatozoa. Both, IVF and embryo transfer are complex techniques susceptible to many external factors. Consequently, there is a greater likelihood of false-negative results. On the other hand, no ideal or easily detectable parameter for an animal-free system of monitoring spermatozoa quality in mice or other species has been identified so far [1,8,9,18,21,31,32]. In our study, possible markers for quality assessment were investigated in detail. Results of the analyses assessing the membrane integrity of cryopreserved spermatozoa presented here were reproducible, and also corresponded with the outcomes of IVF. Identification of a reliable quality control marker allows accurate monitoring in future experiments, possibly by using simpler approaches. It is also important to note that only limited amounts of material are typically available for testing when evaluating samples from GM mice.
One has to take in account that the purpose of cryopreservation of samples from mutant mice is the safe preservation of a GM line so that it can be recovered at a later time. The success of this approach can be influenced by the mutation in the GM line and the inbred genetic background [17]. In contrast, cryopreservation of spermatozoa from livestock is used to distribute samples and to expand herds of the most productive animals [2,19]. Consequently, the markers evaluated for cryopreservation techniques might vary depending upon the purpose of preservation.
Image analysis allowed us to investigate all potential markers of interest. As previously mentioned morphology as determined in this approach is rather weak. Subsequently, it does not qualify for assessment. Motility was examined in parallel with other characteristics of the same sample using a different data acquisition strategy. Concentration of the sperm was found to be only of limited interest. Due to the sampling and cryopreservation protocol we used, concentration of the samples was relatively stable. Furthermore, sample concentration did not significantly influence the quality of the sample. The same observation was previously published [17].
In the present study, we determined that membrane integrity was the most reliable parameter for measuring sample quality. Furthermore, the variance among data obtained from different samples of the same donor mouse and from different sets of fields of view of the same sample was very low.
To validate the strategy we developed, results of the fluorescence microscopy studies were compared to the outcomes of IVF performed with spermatozoa from the same sample. Out of all parameters we evaluated, only membrane integrity significantly correlated with the IVF data. IVF outcomes were categorized as successful if a sufficient number of two-cell embryos were produced and resulted in at least two embryo transfers for recovery the mouse line. The association of membrane integrity with spermatozoa quality was highly correlated even when comparing samples from mice with different genetic background. The outcomes of all analyses were similar.
Several reports have described motility as an important but not determinant parameter for assessing mouse spermatozoa before performing IVF. This agrees with our observations. The importance of membrane integrity becomes more obvious when working with GM mice [17,23,26,27]. GM animals often possess an inbred genetic background. The mutation might negatively influence sperm motility or viability. However, the viability of samples with low motility might still be sufficient for successful recovery of a mutant line.
Since GM donor mice can be re-genotyped and a single IVF procedure is performed to determine the IVF capacity of a line, the technique demonstrated here facilitates reliable quality assessment of cryopreserved spermatozoa. Furthermore, additional animal experiments are not required and only small amounts of material for monitoring purposes would be needed. Our investigation demonstrated that membrane integrity as a reliable quality control marker can be measured with a simple approach by distinguishing PI-negative from PI-positive spermatozoa. Since our method reduces the need for animal experiments, it adheres to the 3R principles postulated by Russell and Burch. Future approaches might simplify this technique.




 XML Download
XML Download