

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Large segmental bone defects caused by events such as trauma, inflammation, or cancer result in serious functional problems, and repair of these injuries is a significant challenge in reconstructive surgery [8,35]. The current gold standard for repairing bone defects is bone graft transplantation despite considerable efforts to develop new therapeutic approaches [22]. This widely-used strategy generally results in a positive clinical outcome, but has many disadvantages including significant morbidity at the donor site, limited supply of bone tissue, increased blood loss, more surgical procedures, decreased osteogenic potential of the grafted bone, potential risk of infection, and problematic immune responses after implantation [8,14]. For these reasons, bone tissue engineering procedures using a combination of mesenchymal stem cells (MSCs) derived from various sources and biomaterials have been investigated as a replacement for more conventional bone graft techniques [7,20].
MSCs are multipotent cells capable of differentiating into various cell types such as osteoblasts, chondrocytes, adipocytes, myoblasts, tenoblasts, and neuronal cells [9,14,30,33]. MSCs have received extensive attention in the fields of tissue engineering and regenerative medicine because they can be easily harvested in a minimally invasive manner, propagate in vitro, and possess valuable properties such as multipotency, paracrine activities, and immune modulation abilities [3,18,30]. In particular, the osteogenic potential of MSCs for repairing bone defects has been evaluated by many researchers [6,25,28,29]. However, careful selection of the most suitable cell source and development of other treatment protocols are needed to achieve more satisfactory bone regeneration results [14].
Currently, it is unclear which type of MSCs is the most suitable for promoting bone regeneration or whether MSCs derived from various tissues have a similar potential for inducing bone formation. Bone marrow (BM) is the most common and readily available cell source for bone engineering procedures, but harvesting this tissue is painful and laborious [8,12,14]. In addition to BM, other tissues including adipose tissue (AT), muscle, umbilical cord blood (UCB), placenta, and Wharton's jelly (WJ) may be sources of MSCs appropriate for bone engineering techniques [3,13,15,27,31].
The purpose of this study was to compare the osteogenic potential of canine MSCs derived from AT, BM, UCB, and WJ. We assessed the cumulative population doubling level (CPDL), mineralization capability, alkaline phosphatase (ALP) activity, and vascular endothelial growth factor (VEGF) production of these MSCs using an in vitro culture assay. The osteogenic ability of the cells was also evaluated using an in vivo orthotopic implantation assay.
Materials and Methods
Animals
Twenty healthy beagle dogs (13 males and 7 females), aged approximately 2 years old with the mean weight of 10.1 ± 1.8 kg were used for the present experiment. The dogs were housed in an indoor facility located in Veterinary Medical Teaching Hospital of Seoul National University, Korea. All animal experimental procedures were approved by the Institutional Animal Care and Use Committee of Seoul National University (SNU-090623-1), Korea.
Isolation and cultivation of canine MSCs
Canine MSCs were obtained from gluteal subcutaneous fat, BM aspirates, WJ, and UCB.
AT was aseptically collected from subcutaneous fat. The fat tissues weighing around 1 g were washed extensively with phosphate-buffered saline (PBS) (Gibco, USA), minced, and digested with collagenase type I (1 mg/mL) at 37℃ for 1 h with intermittent shaking. The suspension was filtered through a 100-µm nylon mesh and centrifuged at 200 × g for 10 min to separate floating adipocytes from stromal cells. Pre-adipocytes in the stromal vascular fraction were plated in T75 flasks (Thermo Fisher Scientific, USA) at a density of 1 × 105 cells/cm2.
BM was aseptically collected from the humeral bone with BM biopsy needle (CareFusion, USA) and 10 mL syringe under anesthesia. The BM was placed in tubes (BD Biosciences, USA) that were treated with an anti-coagulant. The marrow was diluted 1 : 1 with PBS. A Ficoll-Paque plus (Amersham Biosciences, Sweden) density gradient was then used to collect the buffy coat layer. The diluted marrow was gently placed on Ficoll-Paque solution and centrifuged at 400 × g for 20 min. Cells from the buffy coat were washed with PBS and centrifuged at 200 × g for 10 min. The pellets were resuspended in PBS, and the cells were plated in T75 flasks at a density of 1 × 105 cells/cm2.
Fresh canine umbilical cords were obtained after cesarean sections and placed in 20 mL of Hanks' balanced salt solution (HBSS) (Gibco, USA) at 4℃. Following disinfection in 70% ethanol for 30 sec, the umbilical cord vessels were removed while still in HBSS. The mesenchymal tissue (in WJ) was then minced into pieces about 20 mm3 in size and centrifuged at 200 × g for 5 min. After removing the supernatant fraction, the pellet (mesenchymal tissue) was washed with serum-free Dulbecco's modified Eagle's medium (DMEM) (Gibco, USA), and centrifuged at 200 × g for 5 min. The supernatant was aspirated and the mesenchymal tissue was incubated with collagenase type I (1 mg/mL) at 37℃ for 12 h, washed with PBS, and further digested with 2.5% trypsin (Gibco, USA) at 37℃ for 30 min. Fetal bovine serum (FBS) (Hyclone, USA) was then added to the mesenchymal tissue to stop trypsinization. The cells were plated in T75 flasks at a density of 1 × 105 cells/cm2.
Low-density mononuclear cells were isolated from UCB using a Ficoll-Paque density gradient. The diluted UCB was gently placed on Ficoll-Paque solution and centrifuged at 400 × g for 20 min. The cells were washed with PBS and centrifuged at 200 × g for 10 min. The pellets were resuspended in PBS and the cells plated in T75 flasks at a density of 1 × 105 cells/cm2.
All the cells isolated from each type of tissue were incubated overnight in DMEM supplemented with 10% FBS at 37℃ in a humidified atmosphere of 5% CO2. Unattached cells were removed after 24 h by washing with PBS, and cell medium was replaced with fresh medium every 2 days until the cells were confluent. When the confluence was more than 90%, the cells were cryopreserved at -150℃ or subcultured.
Flow cytometry analysis
Trypsinized MSCs were suspended in PBS containing 5% bovine serum albumin (Sigma-Aldrich, USA) at a concentration of 5 × 105 cells/30 µL. The cells were stained with fluorescein isothiocyanate (FITC)-conjugated antibodies specific for CD14 (clone CAM36A; VMRD, USA), CD34 (clone 1H6; Serotec, UK), CD45 (clone CADO18A; VMRD, USA), and CD105 (clone SN6; Serotec, UK) at 4℃ for 30 min. The cells were also incubated with phycoerythrin (PE)-conjugated antibodies against CD44 (clone IM7; Abcam, UK), CD73 (clone 7G2; Abcam, UK), and CD90 (clone DH2A; VMRD, USA) at 4℃ for 30 min. Negative control cells were stained with a FITC-conjugated mouse IgG1 isotype (Invitrogen, USA) or a PE-conjugated mouse IgG1 isotype antibodies (Invitrogen, USA) at 4℃ for 30 min. Expression of the corresponding cell surface markers was evaluated with a FACS Calibur flow cytometer (BD Biosciences, USA) using CELL Quest Pro (BD Biosciences, USA) software.
Comparison of proliferation potential
CPDL was calculated using the formula "χ = {log10(NH)-log10(N1)}/log10(2)" [24] in which N1 is the inoculum cell number and NH is the cell harvest number. To determine the cumulated doubling level, the population doubling level for each passage was calculated and then added to the levels of the previous passages. Since the number of isolated cells from all tissues could be determined for the first time at passage 1, the cumulative doubling number was first calculated for passage 1.
Osteogenic differentiation
Various MSCs from the third passage were cultured in low-glucose DMEM (Sigma-Aldrich, USA) supplemented with 10% FBS in 6-well plates (Thermo Fisher Scientific, USA) at a cell density of 1 × 104/cm2. The DMEM was then replaced with osteogenic medium composed of low-glucose DMEM supplemented with 10% FBS, 0.1 µM dexamethasone (Sigma-Aldrich, USA), 50 µM ascorbic acid-2-phophate (Sigma-Aldrich, USA), and 10 mM beta-glycerophosphate (Sigma-Aldrich, USA), and the cells were incubated for 14 days.
Quantification of mineralization
Alizarin Red S staining was used to observe calcium mineralization. For this procedure, all the different types of MSCs incubated in 6-well plates with osteogenic medium for 14 days were washed twice with distilled water (DW) and fixed in a solution of ice-cold 70% ethanol for 1 h. After being carefully washing five times with DW and then for 15 min with PBS, the cells were stained for 10 min with 40 mM Alizarin Red S (Sigma-Aldrich, USA) at room temperature. Unincorporated dye was removed by aspiration, and the wells were washed four times with 4 mL DW for 5 min with shaking. The plates were then left at an angle for 2 min to remove excess water, re-aspirated, and stored at -20℃ prior to dye extraction. Stained monolayers were visualized by phase contrast microscopy using an inverted microscope (Nikon, Japan). For staining quantification, cells stained with Alizarin Red S were solubilized in cetylpyridinium chloride (1 mL; Sigma-Aldrich, USA) for 1 h as previously described [16]. Absorbance of solubilized Alizarin Red S was measured at 570 nm using a spectrophotometer (SmartSpec 3000 Spectrometer; Bio-Rad, USA).
Measurement of ALP activity
The ALP activity was measured using an ALP assay kit (Takara Bio, Japan) according to the manufacturer's instructions. In brief, a p-nitro-phenyl phosphate (PNPP) solution was prepared by dissolving 24 mg of PNPP substrate in 5 mL ALP buffer. The cells were lysed by adding 500 µL of extraction solution to each well. The cleared supernatant was collected after centrifugation at 16,000 × g at 4℃ for 10 min. The cell lysate supernatant was mixed with 50 µL of PNPP substrate solution and incubated at 37℃ for 30 min. Stop solution (50 µL, 0.9 N NaOH) was added to each well and the absorbance was measured at 405 nm with a spectrophotometer after color development.
Measurement of VEGF
MSCs were seeded in 75 cm2 flasks with DMEM containing 2% FBS and incubated at 37℃ in a humidified atmosphere of 5% CO2. Cultured media was collected after 48 h of incubation. Levels of VEGF in the media were then measured using an enzyme-linked immunosorbent assay kit (Quantikine Canine VEGF; R&D Systems, USA) according to the manufacturer's instructions. The same medium was used as the negative control. The absorbance was measured at 450 nm using a spectrophotometer.
Preparation of the bioceramic scaffold
A β-TCP/poly L-lactide-co-glycolide-co-ε-caprolactone (TCP/PLGC) composite membrane and β-TCP granules were synthesized and provided from the Biomaterial Center, National Institute for Material Science, Japan [7,24]. Briefly, an H3PO4 aqueous solution (5 dm3, 0.67 M) was gradually added to a 1 M Ca(OH)2 suspension with vigorous stirring. pH of the reaction was maintained at 6~7 by the addition of a 28% ammonium aqueous solution. The precipitate was dried at 120℃ and burned at 800℃. The resulting TCP block was crushed into granules. Granules with a particle size between 500 and 1,000 µm were collected by sieve classification. Granules were identified by powder X-ray diffraction (PW-1700 system; Royal Philips Electronics, Netherlands).
PLGC was synthesized by bulk copolymerization of L-lactide, glycolide, and ε-caprolactone using stannous 2-ethylhexanoate. TCP/PLGC composite was synthesized by mixing TCP and PLGC in a weight ratio of 7 : 3 at 180℃ for 10 min. The composite was formed into membranes with a thickness of 200 µm using a hot press (Mini Test Press; Toyo Seiki, Japan) at 180℃.
Orthotopic implantation
The mean diameter of the mid-portion of the radial diaphysis was determined by radiography to create a segmental defect with the critical size in the radial diaphysis. Because the mean diameter of the mid-portion of the radial diaphysis was less than 10 mm, the animals underwent a unilateral resection that created a segmental defect 15-mm long in the radial diaphysis as previously described [2,6]. In brief, the dogs were sedated with intravenous administration of acepromazine maleate (Sedaject; Samwoo Medical, Korea) at a dose of 0.05 mg/kg of body weight. Anesthesia was then induced with the intravenous delivery of 1% propofol (Provive 1%; Claris Lifesciences, India) at a dose of 6 mg/kg of body weight and maintained with isoflurane (Aerrane; Baxter, Canada) in oxygen. Tramadol (Toranzin; Samsung Pharmaceutical, Korea) administered intravenously at a dose of 4 mg/kg of body weight was used as an analgesic. Anesthesia monitor (Datex-Ohmeda; Microvitec Display, UK) was used to monitor physiologic factors including rectal temperature, oxygen saturation, end tidal CO2, electrocardiogram, minimum alveolar concentration value, and pulse rate. Under sterile conditions, a craniomedial incision was made in the skin to expose the diaphysis of the right radius (Fig. 1). An eight-hole, 2.7 dynamic compression plate (Synthes, Switzerland) was contoured and placed on the cranial aspect of the radius. The plate then was removed, and a 15 mm-long segmental defect was made at the mid-portion of the diaphysis using an oscillating saw (Stryker, USA).
The defect was filled with a mixture of β-TCP plus AT-MSCs (AT-MSC group, n = 4), β-TCP plus BM-MSCs (BM-MSC group, n = 4), β-TCP plus UCB-MSCs (UCB-MSC group, n = 4), β-TCP plus WJ-MSCs (WJ-MSC group, n = 4), or β-TCP alone (control group, n = 4). MSCs (1 × 106, third passage) transplanted into each experimental group were suspended in 700 µL of normal saline and mixed with 700 mg β-TCP just before implantation. An appropriate volume of saline was mixed with β-TCP for the control group. The site of implantation was covered with a TCP/PLGC composite membrane. After the plate was reapplied, the fascia and skin were closed. All surgical procedures were performed under the same conditions. Two weeks after surgery, all dogs were able to walk and bear weight using the right antebrachium.
Radiographic examination
Anteroposterior and mediolateral radiographs of the right antebrachium were taken before and immediately after the operation as well as 4, 8, 12, 16, and 20 weeks postoperatively. All of the radiographs were evaluated to identify the presence of osseous union at each host bone-implant interface and monitor changes in implant radiopacity. Bone union at the host bone-implant interfaces was identified by obliteration of the transverse radiolucent line between the host bone and implant that was observed immediately after surgery. The time at which bone healing was observed at the host bone-implant interfaces was also recorded.
Histological and histomorphometric analysis
For histological analysis, the implants of all groups were harvested 20 weeks after the operation. Segments of each radius including the implanted site were excised and fixed in 4% paraformaldehyde. The specimens then were decalcified with 15% formic acid in PBS for 6 to 12 weeks. After decalcification, the segments were dehydrated in a series of ethanol solutions and embedded in paraffin. Longitudinal sections 5-µm thick were cut (Reichert-Jung, Germany) in the coronal plane. These sections were stained with hematoxylin and eosin or azocarmine and aniline blue to observe new bone formation. Stained sections from each group were examined by light microscopy (Olympus, Japan) and photographed using an attached digital camera (Nikon, Japan). For histomorphometric analysis, sizes of the newly formed bone area (NBA) and residual β-TCP area (RTA) were estimated and converted into a percentage of the total implanted area (TA) using an image processing and analysis program (ImageJ; National Institutes of Health, USA).
Statistical analysis
Data were analyzed using SPSS statistical analytical software (ver. 18.0; IBM, USA). A Kruskal-Wallis test was used to assess differences among the groups. A post-hoc test was performed along with a Mann-Whitney U test. p-values less than 0.05 were considered to be statistically significant.
Results
Establishment of canine MSCs from various tissues
Canine MSCs derived from AT, BM, UCB, and WJ were cultured under basal conditions and their cell morphologies at the third passage were compared. Generally, all cells formed a monolayer consisting of spindle-shaped cells (Figs. 2A~D). Fluorescence-activated cell sorting analysis was used to identify cell surface markers on the canine MSCs at the third passage. All cells had the same phenotype and were positive for CD44, CD73, CD90, and CD105 but negative for hematopoietic and endothelial markers including CD14, CD34, and CD45 (Figs. 2E and F).
Proliferation potential of canine MSCs from various tissues
Analysis of the canine MSCs proliferation potential revealed that WJ-MSCs had the lowest population doubling numbers through passages 2~5, but BM-MSCs showed the lowest doubling numbers after passage 7 (Fig. 3). AT-MSCs possessed the highest population doubling numbers at all passages. After passage 6, AT-MSCs had the highest proliferation capacity followed by UCB-MSCs and WJ-MSCs while BM-MSCs had the lowest proliferation potential (Fig. 3).
Comparison of in vitro osteogenic potential
To evaluate the in vitro osteogenic differentiation potential of the MSCs, confluent cultures of AT-MSCs, BM-MSCs, UCB-MSCs, and WJ-MSCs were maintained in osteogenic induction medium for 2 weeks. All the cells generated a large number of mineral nodules within this time. The cells were fixed and stained with Alizarin Red S to visualize the deposition of calcium-rich granules on the cell surface. Intense staining was seen in all MSCs (Fig. 4A). The degree of Alizarin Red S staining was greater in AT-MSCs and UCB-MSCs than BM-MSCs and WJ-MSCs after 2 weeks of subculturing under osteogenic differentiation conditions (Fig. 4B). ALP activity, which is recognized as an early osteoblastic marker, was also measured. Levels of ALP activities of AT-MSCs and UCB-MSCs were significantly greater than those of BM-MSCs and WJ-MSCs (p < 0.05, Fig. 4C).
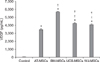
Quantitative analysis of VEGF production
The amount of secreted VEGF, a signal protein produced by cells that stimulate vasculogenesis and angiogenesis, was measured. After 48 h of incubation, VEGF production of the BM-MSCs was significantly greater than that of the other types of MSCs (p < 0.05, Fig. 5). VEGF production of the AT-MSCs was less than that of UCB-MSCs and WJ-MSCs (p < 0.05, Fig. 5).
Radiographic analysis
Representative radiographs for each group of dogs taken at 0, 4, 12, and 20 weeks after surgery are shown in Fig. 6. The β-TCP powder implanted in the bone defects was easily visualized on X-ray images. Over time, implants that had a granular appearance immediately after the operation developed a smoother and more radiopaque appearance in all groups. This was because mineralized tissue had formed in the empty space among the scaffolds as new bone was formed.
A transverse radiolucent line between the host bone and implant was also seen in radiographs taken immediately after the operation. Obliteration of this line was regarded as radiographic evidence of union between the host bone and implant. Radiographic union was observed in seven out of eight host bone-implant interfaces in the BM-MSC group after 20 weeks. Similar results were found in the other experimental groups (AT-MSC group, 6/8; UCB-MSC group, 7/8; and WJ-MSC group, 7/8). In contrast, union between the host bone and implant was seen at only one out of eight interfaces in the control group after 20 weeks. The times at which osseous union at the host bone-implant interfaces appeared were similar among the experimental groups (Table 1). The control group was excluded from calculating this time because radiographic union was observed at only one interface. The time of the one union observed in the control group was 16 weeks.
Histological and histomorphometric analysis
In all experimental groups, substantial bone formation was observed throughout the bone defect (Fig. 7). Newly formed bone was primarily observed on the surface of intact β-TCP particles. In addition, cortical continuity was identified by the lack of a distinct boundary between host bone and newly formed bone in these experimental groups. Newly woven and lamellar bones in direct contact with the β-TCP particles and osteocytes embedded within the bone matrix were observed at higher magnification (×200). Hematopoiesis was also observed within the bone matrix. Overall, the histological findings were similar among the experimental groups. In contrast, the control group treated with β-TCP alone had minimal new bone formation throughout the implant, and continuity between native bone and the implant was absent.
The bone formation capacity was assessed in all groups by measuring areas of newly formed bone with histomorphometry. Percentages of NBA to TA were significantly higher for the experimental groups treated with the canine MSCs from various sources compared to the control group (Table 2, p < 0.05). However, the amounts of new bone formed were not significantly different among the experimental groups. The RTA to TA percentages for all groups were not significantly different.
Discussion
For the therapeutic use of MSC-based bone tissue engineering, it is essential to identify suitable conditions to promote osteogenesis. In the present study, we focused on MSCs from different sources to establish suitable conditions for bone repair. A few reports have recently compared the osteogenic abilities of MSCs derived from different tissues [11,19]. However, these studies evaluated osteogenesis only by performing in vitro assays or in vivo ectopic implantation. In vitro osteogenesis or in vivo ectopic bone formation may not represent the actual osteogenic potential of MSCs transplanted into a bone defect. Therefore, the current study used not only an in vitro assay but also in vivo orthotopic implantation to investigate bone formation by various types of MSCs.
Our in vitro osteogenic study showed that AT-MSCs and UCB-MSCs had a greater osteogenic potential than BM-MSCs and WJ-MSCs although subcultures of all types of MSCs formed sufficient bone matrix under osteogenic differentiation conditions. However, all MSCs induced substantial in vivo bone formation and significant differences in the levels of bone formation promoted by the various MSCs were not observed. These findings indicate the osteogenic potential observed in vitro and in vivo can be slightly different for each type of MSCs. This hypothesis is supported by previous reports demonstrating that findings from an in vitro osteogenic differentiation assay correlate poorly with the results of an in vivo osteogenic differentiation assay [5,11]. These reports also suggested that confirming osteogenic potential using an in vivo rather than an in vitro system is more appropriate to ascertain bone regeneration abilities.
Differences of osteogenic potential as measured by the in vitro and in vivo experiments can be potentially explained by several factors. First, vascularization may have contributed to homeostasis of the microenvironment that promoted MSC survival and bone formation. Some reports proposed that the initial presence of MSCs stimulates bone formation by inducing vascularization [1,7,21]. MSCs are generally known to secrete substantial quantities of VEGF that plays a central role in the angiogenic response. Our present study measured VEGF production in vitro to determine the ability of each MSC type to promote neovascularization. BM-MSCs secreted significantly greater quantities of VEGF compared to other MSCs while AT-MSCs produced the lowest amount of VEGF. In contrast, bone matrix deposits formed by AT-MSCs under osteogenic conditions were larger than those of other MSCs, and BM-MSCs had a weaker osteogenic differentiation capability compared to AT-MSCs and UCB-MSCs. However, new bone was formed in a similar manner by all types of MSCs in the in vivo osteogenic assay. Based on these results, we can speculate that in vivo bone formation may be affected by the ability of MSCs to facilitate neovascularization. Indeed, hematopoietic tissues were observed more frequently within the bone matrix in histological sections from all experimental groups compared to the control group.
MSCs may also affect bone formation by releasing factors that stimulated the induction and migration of cells in the surrounding bone [7,26]. In reality, all bone healing process cannot be directly influenced by osteogenic differentiation of the transplanted cells. According to a previous report that investigated a transplantation model of live bone grafts from Rosa26A mice [36], only 70% of the induced osteogenesis on the graft depended on the expansion and differentiation of the donor progenitor cells. Furthermore, a previous study from our laboratory revealed that significant levels of cytokines released by canine UCB-MSCs 1 day after implantation can enhance bone regeneration [7].
Finally, interactions between MSCs and extracellular microenvironments may influence the fate of the cells. Local tissue microenvironments can affect stem cell attachment and migration, presentation of chemical and physical cues, and the binding of soluble factors [17,34]. These three different possibilities may have caused all types of MSCs to have similar levels of osteogenic potential in vivo and different degrees of osteogenic potential in vitro.
In the present study, allogenic MSCs were used to regenerate bone in long-bone defects with critical sizes. This may raise the issue of a possible adverse reaction or eventual rejection of the allogenic MSCs. However, previous studies have indicated that MSCs may be immune-privileged cells [2,4]. A previous in vivo orthotopic investigation demonstrated that allogenic MSCs do not provoke adverse immune responses, and are present in newly forming bone tissues when implanted into critical-sized bone defects in dogs without the use of immunosuppressive therapies [2]. In the present study, we showed that allogenic MSCs could enhance bone regeneration in critical-sized canine segmental defects without immunosuppressive therapies. The amounts of bone formed throughout the implants loaded with allogenic MSCs were significantly greater than those formed with cell-free implants. Thus, our study demonstrated the potential clinical application of allogenic MSCs for bone repair procedures.
BM-MSCs have been mainly used with biomaterials as a cell source for bone engineering. However, BM harvesting is a laborious and traumatic procedure, and the number and differentiation potential of the MSCs decrease with increasing age of the MSC donor [23]. Therefore, interest in the possibility of obtaining MSCs from other tissue sources has increased. In our study, AT, UCB, and WJ were used to obtain MSCs, and we measured the osteogenic potential of these cells to determine if they can be used as alternatives for BM-MSCs. The main advantages of AT-MSCs are that these cells are easy to obtain, are available in substantial quantities, and can be harvested with no risk of donor site morbidity. In addition, AT-MSCs grow stably in vitro and have a high expansion rate [32]. AT-MSCs were also shown in the present study to have a higher proliferation potential than the other types of MSCs. UCB-MSCs and WJ-MSCs can also be harvested painlessly without the risk of patient morbidity, maintain stemness in vitro, and are immune-privileged cells with surface characteristics capable of overcoming rejection [15,27]. In addition, UCB-MSCs have a significantly greater osteogenic potential in vitro compared to BM-MSCs according to a previous report [10]. Similarly, results of the in vitro assays performed in the present study indicated that UCB-MSCs possessed a greater osteogenic potential than BM-MSCs.
In conclusion, we demonstrated that AT-MSCs, UCB-MSCs, and WJ-MSCs have many advantages that make these cells ideal for bone repair procedures. These cells also possess a degree of osteogenic potential similar to that of BM-MSCs. Our findings suggest that AT-MSCs, UCB-MSCs, and WJ-MSCs are promising types of stem cells for bone regeneration techniques and can be used in place of BM-MSCs.








 XML Download
XML Download