

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Ischemic heart disease is due to deficient coronary circulation. This pathologic condition is usually associated with myocardial damage caused by cardiac ischemia-reperfusion (I/R) and has been the leading cause of death worldwide [35]. Ischemic preconditioning (IPC) achieved by repeated short episodes of ischemia protects the heart against a subsequent sustained period of ischemia, and consequently reduces post-ischemic arrhythmia, abnormal contractility, and the extent of myocardial infarction [5].
IPC also can be mimicked by pharmacological pretreatment [20], heat shock [16], and mechanical stretching of the heart [28]. Many studies have shown that stretch preconditioning (SPC) enables the myocardium to better tolerate a subsequent ischemic insult. Ovize et al. [28] showed that brief stretching of the myocardium significantly reduces myocardial infarction extent in canines, and that this protective effect is blocked by gadolinium (Gd3+), a potent inhibitor of stretch-activated ion channels (SACs). Additionally, temporary myocardial stretching induced by increased diastolic pressure in the left ventricle results in significantly decreased infarct size and improves post-ischemic contractile function in isolated rat hearts [25,27]. However, details about the signaling pathways involved in stretch-induced cardioprotection (SIC) have not been previously reported.
Mechanical stretching directly affects cardiac functions and rapidly activates multiple signal transduction factors in cardiac myocytes such as the c-Jun N-terminal kinase, mitogen-activated protein kinases, protein kinase C, and Janus kinase/signal transducers and activators of transcription pathways [6,29]. The serine/threonine-specific protein kinase Akt (also known as protein kinase B), is a potent survival factor [7] and one of the primary kinases activated by phosphatidylinositol-3-kinase (PI3K) which is activated by several myocardial protective ligand-receptor systems including insulin [3], insulin-like growth factor-1 [22], and gp130 signaling pathways [24]. Researchers have also shown that cardiac stretching can induce Akt signaling [18,34]. Activated Akt promotes cell survival by phosphorylating and inactivating glycogen synthase kinase-3β (GSK-3β), the pro-apoptotic Bcl-2 family member BAD [1], and caspase-9 [4]. Nuclear factor-κB (NF-κB) is also activated by Akt, resulting in the transcription of pro-survival genes [32]. As a result, activated Akt substantially reduces infarction size and dramatically improves post-ischemic myocardial function [8]. Interestingly, there is a possibility that SIC might share a common pathway with IPC in the heart [9], and several groups have reported that Akt is likely to be involved in a common signaling pathway affecting both IPC- and heat shock-induced cardioprotection [15,39].
SACs are proposed mechanotransducers which conduct ionic currents in response to membrane tension such as mechanical stretching [21]. Kim et al. [18] previously reported that PI3K-dependent phosphorylation of Akt is increased by mechanically stretching the heart, and stretch-mediated increase of vascular endothelial growth factor production is suppressed by Gd3+. These findings suggest that SACs play an important role as mechanotransducers in rat heart and these channels might be associated with the PI3K/Akt signaling pathway.
In the present study, we examined the possibility that myocardial stretching enables the myocardium to better tolerate I/R damage. We also evaluated the underlying mechanism of SIC to determine whether this type of cardioprotection is related with Akt activation through the SACs. Our results suggest that SACs and PI3K/Akt pathway dependent GSK-3β inhibition plays an important role in the cardioprotection induced by transient stretching preconditioning.
Materials and Methods
Ex vivo model of cardiac stretching
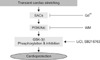
All animal experiments were conduced in accordance with the National Institutes of Health (USA) Guidelines for the Care and Use of Laboratory Animals and were approved by the Chungbuk National University Medical School Research Institutional Animal Care and Use Committee (Korea). Specific pathogen-free male Sprague-Dawley (7 weeks old, 200~220 g; Koatech, Korea) rats were anesthetized with Zoletil (30 mg/kg; Virbac, France) and xylazine (10 mg/kg; Bayer, Germany). Hearts were excised and then perfused at a constant pressure (perfusion pressure was maintained at 80 cmH2O) in a non-recirculating Langendorff mode with Krebs-Henseleit buffer (in mmol/L: 118 NaCl, 4.7 KCl, 1.25 CaCl2, 1.2 MgSO2, 10 glucose, 25 NaHCO3, and 1.2 KH2PO4) saturated with a mixture of 95% O2/5% CO2 at 37℃.
To stretch the left ventricle of the isolated hearts, a plastic catheter with a small balloon tip (made in our laboratory) was inserted into the left ventricle through the mitral valve. The left ventricle was subjected to stretching for 5 min by expanding the inserted balloon to raise the left ventricular end-diastolic pressure (LVEDP) to 40 mmHg. To induce I/R injury to the heart, isolated rat heart was subjected to global ischemia for 30 min followed by reperfusion for 60 min (Fig. 1A). Before undergoing sustained ischemia, the hearts were assigned to different groups (n = 6~10) that underwent 30-min "pretreatments" consisting of: 1) no treatment (the I/R control group), 2) three cycles of 5-min ischemic periods (the IPC group), 3) 5 min of stretching (the SPC group), 4) 10 min of lithium chloride (final concentration of 3 mM in Krebs-Henseleit buffer; Sigma, USA) or SB216763 (3 µM; Tocris Cookson, UK) treatment (the GSK-3β inhibitor group), 5) treatment with wortmannin (3 µM; Sigma, USA) and 5 min of stretching (PI3K inhibitor group), and 6) treatment with Gd3+ (10 µM; Sigma, USA) and 5 min of stretching (the SAC inhibitor group).
In vivo model of cardiac stretching
To induce mechanical stretching in the rat myocardium in vivo, aorto-caval shunt (ACS) surgery was performed as previously described [18]. Rats were anesthetized by intraperitoneal administration of ketamine (80 mg/kg; Bayer, Germany) and xylazine (5 mg/kg; Bayer, Germany). A shunt between the inferior vena cava and abdominal aorta was created using an 18-gauge needle at a location 5 mm caudal to the left renal vein. Patency of the shunt was verified by the mixing of arterial and venous blood. Sham operations were performed in control rats in an identical manner except that the needle was not inserted. The abdominal incision was closed with sutures and the animals were allowed to recover.
Central venous pressure (CVP) and LVEDP were monitored using catheters inserted into the superior vena cava and left ventricular lumen as previously reported [26]. CVP and LVEDP were recorded using a computer data acquisition system (model ML870 PowerLab 8/30; AD Instruments, Australia). For the in vivo stretching experiment, the rats were randomly divided to four groups of 6~10 rats each (Fig. 1B). Group 1 did not undergo any intervention (the control group). Group 2 received a brief volume overload in the left ventricle through the ACS for 5 or 30 min. Group 3 received an intravenous infusion of wortmannin (0.6 mg/kg), and 15 min later also received a brief volume overload through the ACS similar to group 2. Group 4 received an infusion of Gd3+ (16 mg/kg), and 15 min later was subjected to the ACS similar to group 2. Infusion was performed for 5 min.
Evaluation of infarct size
Infarct size was measured as previously described [19]. The hearts were cut into six transverse sections parallel to the atrioventricular groove, and incubated in a 1% solution of 2,3,5-triphenyltetrazolium chloride in phosphate buffer for 10 min at 37℃. The sections were photographed using a PowerShot A640 digital camera (Canon, Japan) and the images were traced to identify the boundaries of the infarct area with Photoshop CS6 (Adobe, USA). Finally, the normalized percent infarct area was calculated by dividing the total infarct size by the total heart volume.
Measurement of cardiac functional recovery
Left ventricular pressure was monitored as previously described [19]. Functional recovery of the heart was evaluated by comparing pre- and post-ischemic functional indices. The functional index was calculated as by multiplying the heart rate (HR) by the left ventricular developed pressure (LVDP; systolic pressure minus the end-diastolic pressure in mmHg).
Measurement of lactate concentration
Samples of coronary venous effluent were collected periodically from control and SPC hearts and immediately frozen in liquid nitrogen and stored in -78℃. Measurement of lactate concentration was performed by enzymatic assay (Lactate Reagent; Sigma, USA) according to manufacturer's protocol.
Immunoblot analyses of Akt and GSK-3β
Myocardial tissues excised from each heart were homogenized (WiseStir; DAIHAN Scientific, Korea) in ice-cold lysis buffer (50 mmol/L Tris, pH 7.4; 1 mmol/L EDTA, 0.5 mmol/L DTT, 0.4 mmol/L PMSF, 1 mmol/L sodium vanadate, 1 mmol/L sodium orthovanadate, and 5 mmol/L sodium fluoride) containing a protease inhibitor cocktail (Sigma, USA), and subjected to immunoblot analysis as previously described [18]. Twenty µg of each lysate were separated by 12% SDS-polyacrylamide gels and transferred to immobilon-polyvinylidine fluoride membranes (Millipore, USA). The membranes were blocked with 5% skim milk and Tris-buffered saline for 1 h at 25℃. Next, the membranes were incubated overnight with mouse monoclonal anti-phospho-Akt and anti-phospho-GSK-3β antibodies (1 : 1,000; Cell Signaling Technology, USA), or anti-Akt and anti-GSK-3β antibodies (1 : 1,000; Cell Signaling Technology, USA) in 5% skim milk and Tris-buffered saline at 4℃. After washing three times with Tris-buffered saline (15 min per wash), the blots were incubated with a goat anti-rat IgG antiserum conjugated to horseradish peroxidase (1 : 1,000; Santa Cruz Biotechnology, USA) for 2 h at 25℃. The blots were washed three times with Tris-buffered saline and antibody binding was detected with an enhanced chemiluminescence plus detection system (Santa Cruz Biotechnology, USA).
Statistical analysis
Results were analyzed with the SPSS statistical package (ver. 12.0; IBM, USA) and are presented as the mean ± SE. The data were compared using Tukey's honestly significant difference test and a one-way analysis of variance (ANOVA). p-values < 0.05 were considered statistically significant.
Results
Protective effects of stretching on the heart ex vivo
The protective effects of stretching against I/R on the rat hearts ex vivo were evaluated according to changes in the normalized infarct size and recovery of cardiac function after I/R (Fig. 2A). IPC or SPC significantly decreased infarct size (3.7 ± 1.5% and 15.9 ± 2.2%, respectively) compared to the control hearts (39.1 ± 2.2%). There was no significant change in infarct size in the control hearts treated with wortmannin or Gd3+. However, treatment with these two compounds significantly diminished the ability of SPC to limit infarct size (34.2 ± 2.7% and 33.0 ± 2.8%, respectively). Lithium chloride or SB216763 treatment instead of SPC reduced the infarct size to 24.8 ± 2.3% and 19.2 ± 2.5%, respectively.
To assess the recovery of heart function after ischemia, four functional parameters were evaluated: functional recovery, LVEDP, HR, and LVDP. Recovery of post-ischemic cardiac function was significantly improved in the IPC and SPC groups (31.5 ± 3.9%) compared to the control group (4.6 ± 0.61%, Fig. 2B). Lithium chloride or SB216763 treatment alone also resulted in a level of functional recovery similar to that produced by SPC. Even with SPC, wortmannin or Gd3+ treatment reduced post-ischemic functional recovery similar to that observed in the I/R control group. Recovery of post-ischemic LVEDP, HR, and LVDP also showed similar patterns. These results suggest that transient mechanical stretching of the myocardium effectively conferred protection against I/R injury in rat heart.
Stretch-induced Akt activation in the heart ex vivo
Thirty min of sustained ischemia significantly decreased the levels of Akt phosphorylation and resulted in a corresponding decrease of GSK-3β phosphorylation in the I/R control group. SPC produced a 2-fold increase in the levels of Akt phosphorylation before ischemia and GSK-3β phosphorylation after 30 min of ischemia. Wortmannin treatment markedly blocked both SPC-induced Akt and GSK-3β phosphorylation (Figs. 3A and C). Gd3+, a SAC blocker, completely abolished SPC-induced increases in Akt phosphorylation (Figs. 3B and C). Consequently, Gd3+ also decreased GSK-3β phosphorylation to the control baseline level. These results suggested that SACs might be required to activate Akt in the heart during SPC. Our findings also indicated that mechanical stretching can induce Akt phosphorylation, probably through SACs, and that Akt activation via the PI3K pathway and/or GSK-3β phosphorylation might be involved with SPC-induced cardioprotection.
Measurement of hypoxic stress in stretched myocardium
Time-matched lactate concentrations did not vary when comparing the normal control and stretched hearts. These results suggested that mechanical stretching per se was not responsible for inducing ischemic insult in the isolated hearts (Table 1).
Stretch-induced Akt activation in the heart in vivo
In the present study, we stretched the left ventricle of the heart in vivo by pressure overload via the ACS. Two ACS-associated parameters were examined: hemodynamic changes and induction of ischemic cardiac insult. After the ACS was created, CVP increased 2-fold and LVEDP was elevated 3-fold (data not shown). Measurements taken at different time points after ACS creation (n = 6, 5 min to 1 h) did not reveal any significant changes in the elevated CVP and LVEDP, confirming that the ACS adequately stretched the left ventricle.
Akt phosphorylation due to the ACS was initiated as early as 20 min and sustained for 30 min (Fig. 4), which indicated that not only ex vivo SPC but also in vivo cardiac stretching by the ACS can activate Akt. In contrast, the time course of ACS-induced Akt phosphorylation was different from that observed in the isolated control hearts. In the ACS group, more time was required for Akt phosphorylation than in the ex vivo stretch model. Consistent with the results from the hearts stretched ex vivo, wortmannin and Gd3+ pretreatment significantly inhibited the Akt phosphorylation increased by the ACS.
Protective effects of stretching on the hearts in vivo
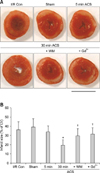
Myocardial infarct sizes of the control and sham-operated group expressed as a percentage of the area at risk were 36.2 ± 8.5% and 39.2 ± 8.9%, respectively. In order to confirm the positive relationship between Akt activation and the cardioprotective effect of stretching, we measured infarct size in the hearts that underwent different periods of hemodynamic overload (5 and 30 min) induced by the ACS. In the 5-min ACS group in which hemodynamic overload was applied for 5 min in the hearts, infarct size expressed as a percentage of the area at risk was 33.8 ± 6.5%. This figure was not significantly different from those observed in the hearts from control and sham-operated rats. On the other hand, hearts subjected to 30 min of hemodynamic overload via the ACS (Fig. 5) were quite resistant to I/R injury and showed significantly reduced infarct size (19.6 ± 5.6%). These results showed that differences in the level of Akt phosphorylation observed between the groups treated for 30 min with the ACS and the other experimental groups (Fig. 4) may partially account for the protective effect of myocardial stretching. Additionally, treatment with wortmannin or Gd3+ completely abolished the cardioprotective effect of the ACS, confirming that Akt activation (which might be related to GSK-3β inhibition) plays a critical role in stretch-induced protection against I/R insult (Fig. 6).
Discussion
This study demonstrated that Akt phosphorylation in rat myocardium was induced during an early phase of mechanical stretching both ex vivo and in vivo, and that this event was followed by the phosphorylation of GSK-3β. Brief mechanical stretching of the left ventricle preconditioned rat heart and offered protection against I/R injury, thereby reducing myocardial infarct size and improving post-ischemic cardiac function. PI3K-dependent Akt phosphorylation was suppressed by wortmannin or Gd3+. Likewise, the effects of mechanical SIC were also abolished by the administration of wortmannin or Gd3+. Cardioprotection was induced by streching in both the ex vivo and in vivo models by pretreatment of two GSK-3β inhibitors, lithium chloride or SB216763, suggesting that GSK-3β inactivation might mediate cardioprotection. These results indicate that PI3K-dependent Akt phosphorylation and subsequent GSK-3β inhibition play an important role in the cardioprotection conferred by transient stretching preconditioning, and that this is regulated by SACs.
Repeated short episodes of coronary artery occlusion render the heart more resistant to a subsequent sustained period of ischemia and reduce the extent of I/R injury. This phenomenon is termed IPC. IPC can be mimicked by non-ischemic stimuli including pharmacological or mechanical stimulation. Numerous studies have demonstrated that mechanical stretching of the myocardium can precondition the heart and render this organ more resistant to the deleterious effect of prolonged ischemia. It has also been shown that brief stretching of the left ventricle by increasing LVEDP results in tolerance to subsequent I/R damage in a manner similar to that associated with classical IPC [6,25,27,29]. Furthermore, this stretch-mediated cardioprotection is abolished by Gd3+, a blocker of SACs. Although many studies of the protective effects of SPC have been performed, the exact underlying mechanisms have not yet been elucidated. In the present study, we found that PI3K-dependent Akt phosphorylation occurred in the myocardium with mechanical stretching, suggesting the possible involvement of the PI3K/Akt pathway in the cardioprotective effect of SPC. In accordance with previous reports [6,25,27,29], we found that the SPC effects were abolished by Gd3+, indicating that stretch-induced Akt activation and cardioprotection is linked to SACs which act as mechnotransducers.
PI3Ks are a family of enzymes involved in diverse cellular functions including cell survival, growth, and proliferation. These kinases are reported to be involved in the insulin signaling pathway as well as IPC [37]. Many functions of PI3Ks are related to Akt phosphorylation and GSK-3β inactivation. It has been found that overexpression of mutant GSK-3β abolishes the cell survival effect of PI3K [30]. Moreover, Ma et al. [23] demonstrated the role of the PI3K/Akt signaling pathway in cardioprotection induced by ischemic postconditioning in minipigs.
Akt, also known as protein kinase B, regulates cell survival and metabolism by binding to and activating (or inactivating) its downstream substrates through phosphorylation [33]. BAD, a pro-apoptotic protein belonging to the Bcl-2 family, can be phosphorylated on Ser 136 by activated Akt and consequently loses its pro-apoptotic function. Inactivated BAD is responsible for mitochondrial membrane stabilization, resulting in the reduced release of proapoptotic proteins from mitochondria [12]. Caspase-9 may also be inactivated by Akt, which results in inhibition of the apoptotic pathway [40]. Additionally, Akt can activate NF-κB, thereby increasing the transcription of pro-survival genes. In this study, we did not examine the role of BAD, caspase-9, or NF-κB and focused on the role of GSK-3β in cardioprotection provided by myocardial stretching. BAD, caspase-9, and NF-κB could be other effector molecules responsible for SIC. This possibility remains to be clarified.
In the present study, we confirmed the role of Akt in SIC. Myocardial stretching also increased in vivo Akt phosphorylation in a time-dependent manner. In myocardium mechanically stretched by the ACS, Akt phosphorylation was significantly increased after 30 min (but not 5 min) of ACS placement. Concurrent with the changes observed in Akt activity, reduced infarct size was only observed in the 30-min ACS group. Based on these results, phosphorylation of Akt is likely to be responsible for the resistance to ischemia induced by myocardial stretching.
In our study, it is likely that GSK-3β phosphorylation in response to myocardial stretching was not concordant with Akt activation. Phosphorylation of GSK-3β was increased after 30 min of ischemia at which time the levels of phosphorylated Akt had already decreased. These results suggested the possibility that GSK-3β is not a downstream effector of Akt in stretched heart tissues. However, this discrepancy may be explained by accumulation of phosphorylated GSK-3β in response to Akt activation even if Akt phosphorylation gradually decreases during the ischemic period. Akt has been identified as an upstream regulator of GSK-3β. Since Akt is a major kinase downstream from PI3K, many stimuli that activate PI3K, such as endothelin-1, Fas, and pressure overload, also inhibit GSK-3β through Akt [2,10]. In the present study, phosphorylation of Akt and GSK-3β significantly increased with mechanical stretching. This was observably attenuated by wortmannin, a PI3K inhibitor, which suggested that mechanical stretching leads to GSK-3β phosphorylation and inhibition, possibly through the PI3K/Akt pathway. Moreover, GSK inhibitors (lithium or SB216763) mimicked the protective effects of SIC when used to treat hearts prior to the induction of ischemia. Our results indicated that phosphorylation and subsequent inactivation of GSK-3β through the PI3K/Akt pathway play an important role in the cardiac protection afforded by SPC. Although GSK-3β is an established target of Akt, it is unlikely that regulation of GSK-3β is solely dependent on Akt in stretched rat heart. Integrin-linked kinase, protein kinase A, protein kinase C, p90 ribosomal S6 kinase, and p70S6K are other upstream kinases that possibly regulate GSK-3β [11]. Further investigation will be necessary to elucidate the precise mechanisms underlying GSK-3β phosphorylation during SPC.
On the other hand, acute volume overload might increase LVEDP and workload of the heart, and disturb coronary blood flow. Under these conditions, acute volume overload can result in an imbalance between oxygen supply and demand in the stretched myocardium and thereby induce IPC. Huang et al. [13] recently reported that myocardial stretching induced by brief pressure overload protects rabbit heart against ischemic injury. However, they did not examine the possibility of ischemia produced by acute pressure overload. To investigate this possibility, myocardial lactate concentrations were measured in coronary effluent from isolated rat hearts in the present study. Lactate levels increased after ischemia in control hearts but not after mechanical stretching, suggesting that myocardial stretching did not induce myocardial hypoxia or ischemia. However, we did not measure coronary blood flow in our study and therefore cannot rule out the possibility that blood flow redistribution results in subendocardial ischemia in stretched rat heart in vivo.
Remote preconditioning in the heart (termed intracardiac preconditioning) was first performed in a study by Przyklenk et al. [31]. Short occlusion of the circumflex artery was found to protect the myocardium supplied by the left anterior descending coronary artery. Kim et al. [18] previously observed increased diastolic wall stress in the ischemic heart as well as increased atrial natriuretic peptide mRNA expression that is known to be induced by myocardial stretching in non-ischemic myocardium of regionally ischemic rat heart. Intracardiac remote preconditioning has been widely documented in various animal models [31,36]. Effects of this preconditioning are thought to be due to the release of humoral factors or activation of nerve pathway [36], but the precise underlying mechanism remains to be elucidated. In the present study, we found that PI3K/Akt pathway-dependent GSK-3β inhibition plays an important role in the cardioprotection induced by transient stretching preconditioning. To our knowledge, this is the first report suggesting a possible role of this pathway in the protective effect of intracardiac remote preconditioning.
Previously reported results of IPC in cardiac surgery are conflicting. Although some researchers did not observe any benefit of IPC [17], most studies have shown the protective effects of IPC in cardiac surgery [14,38]. However, the possibility of embolism development in the ascending aorta has impeded the widespread use of IPC in clinical settings. In our study, preconditioning performed by volume overloading the heart was unaccompanied by ischemia. Therefore, a lower risk of embolism formation could be expected when using this technique. Although SPC cannot be used for patient treatment per se, mechanical stretching of the left ventricle represents a novel non-ischemic technique for providing myocardial protection. Additionally, the underlying mechanism of SPC may facilitate the development of new therapies for treating patients with ischemic heart disease.





 XML Download
XML Download