

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Ehrlichia (E.) canis, which is the primary etiologic agent of canine monocytic ehrlichiosis (CME), is a Gram-negative obligate intracellular bacterium that replicates in monocytes and macrophages [5]. E. canis is primarily transmitted by the brown dog tick Rhipicephalus sanguineus [2]. CME has been reported throughout the world, with a higher frequency in tropical and subtropical regions [12]. Dogs infected with E. canis can present with a wide spectrum of symptoms, ranging from subclinical infection to death [1,6,11]. Clinical signs often include anorexia, dyspnea, hyperthermia, lethargy, weight loss, and bleeding disorders [3,4,22].
Although the genome of the E. canis prototype strain Jake was completely sequenced in 2006 [8], the genetic diversity of E. canis strains worldwide is still poorly defined. To date, most molecular epidemiology studies of E. canis have focused on the 16S ribosomal RNA gene (16S rDNA), while much less is known about the other genes. Unfortunately, molecular characterization of the 16S rDNA has provided little information regarding strain diversity and suggests a high level of conservation [16,20,21]. It has been reported that the 16S rDNA partial sequences were 99.9~100% identical among isolates of E. canis from South America (VHE and VDE isolates), North America (Oklahoma isolate), Asia (Thai, Chinese, and Japanese Kagoshima isolates), and the Middle East (Turkish and Israeli isolates) [15,23]. These observations indicated that the 16S rDNA sequence might not be the best target for evaluation of the genetic diversity of E. canis.
Zhang et al. [24] recently analyzed and compared the gp200 gene sequences of E. canis isolates from the United States, Brazil and Israel and found that gp200 was highly conserved between the isolates from the USA and Brazil, but that substantial diversity was present in the Israeli isolate. The gp200 gene encodes the largest major immunoreactive protein that has been identified in E. canis to date [10,14]. The gp200 protein has been shown to be especially sensitive to immunodiagnositic antigen for E. canis infections and to provide species specificity [9]. Investigation of the extent of sequence variation in this antigen candidate may help elucidate the diversity of E. canis isolates and pave the way for development of an effective vaccine for CME. Herein, we analyzed the sequence variations in the gp200 gene of the E. canis parasite from naturally infected dogs in Taiwan. To define the genotype of the organism, the 16S rDNA was also analyzed.
Materials and Methods
Blood samples and microscopic examination
Eighty-seven blood samples were collected from dogs exhibiting clinical signs compatible with ehrlichiosis that presented to the Veterinary Teaching Hospital of National Chung Hsing University and eight veterinary clinics throughout Taiwan during 2009. Blood was collected into sterile tubes containing anticoagulant (EDTA) and then stored at 4℃ until arrival at the laboratory for further processing. Giemsa-stained blood smears were examined by light microscopy (by observing 100 microscopic fields at 1,000× magnification) for the presence of morulae or inclusion bodies of E. canis. Genomic DNA was extracted from 200 µL of whole blood samples using a QIAamp DNA Blood Mini Kit (Qiagen, USA) according to the manufacturer's protocol. Nucleic acid was eluted into 100 µL of elution buffer and stored at -20℃ until further use.
PCR amplification
The oligonucleotide primers used for the amplification of E. canis genes (16S rDNA and gp200) were designed using primer design software (PrimerSelect; DNAStar, USA) and information from the E. canis genome (GenBank accession number CP000107) [8]. All primers used in this study are shown in Table 1. For each PCR amplification, 5 µL of extracted DNA was used as the template in a 25 µL reaction mixture containing 1× PCR buffer (Promega, USA), 2.5 mM MgCl2, 0.2 mM deoxynucleotide triphosphates (dNTPs), 0.8 µM of each primer and 0.125U TaqBead Hot Start Polymerase (Promega, USA). The reactions were conducted in an ABI2700 thermocycler (Applied Biosystems, USA) according to the following parameters: 94℃ for 3 min followed by 35 cycles of 94℃ for 1 min, 58℃ for 1 min and 72℃ for 1.5 min, followed by an extension step at 72℃ for 5 min.
Cloning and sequencing
The resulting PCR products were electrophoresed on a 1.2% agarose gel that was stained with ethidium bromide to check the size of the amplified fragments by comparison to a DNA molecular weight marker (100-bp DNA Ladder; Promega, USA). In each case, the single amplified product of the expected size was column purified using the QIAquick PCR Purification Kit (Qiagen, USA) and then ligated into the pGEM-T vector (Promega, USA) for subsequent transformation in Escherichia coli DH5α competent cells. One plasmid vector containing the insert was purified from each clone using the QIAprep Spin Miniprep Kit (Qiagen, USA), after which it was sequenced using an ABI PRISM 3730 capillary sequencer (Applied Biosystems, USA) and the Dye Terminator Cycler Sequencing Kit (Applied Biosystems, USA) with the T7 or SP6 vector primer. Both the sense and antisense strands of each PCR-amplified product were sequenced, and the sequences were then manually edited to resolve ambiguities. A consensus sequence was obtained for each amplified PCR product by comparing both the sense and antisense sequences.
Sequence and phylogenetic analyses
The BLAST program (NCBI, USA) was used for comparison and analysis of sequence data obtained in this study versus those previously deposited in the GenBank database. Multiple sequence alignment was conducted using Clustal W version 1.8 [18]. Phylogenetic trees were inferred using the neighbor-joining method as implemented by the MEGA software version 4 [17]. The distance matrix of amino acid divergences was calculated according to Kimura's two-parameter model furnished by MEGA. A bootstrap resampling technique of 1,000 replications was conducted to statistically support the reliabilities of the nodes on the trees.
Results
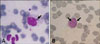
The morulae or inclusion bodies of E. canis were detected by microscopic examination in 17 out of 87 blood smears (19.6%) of dogs exhibiting clinical signs compatible with ehrlichiosis (Fig. 1). Positive blood samples from these parasitemic dogs were subjected to DNA extraction, amplification and sequencing. All 16S rDNA sequences of 1,620 bp derived from 17 samples were found to be identical to each other. The representative sequence obtained from this study has been deposited in the GenBank database under accession number GU810149. The sequence had 100% identity with the corresponding sequences from E. canis isolates in Brazil (GenBank accession numbers EF195134), Greece (EF011110-1), Italy (EU439944), Turkey (AY621071), Venezuela (AF373612-3), and Thailand (EU263991 and EF139458), 99.9% homology with a Japanese isolate (AF536827) and a strain from the USA (M73221), 99.8% identity with a Chinese isolate (AF162860), 99.7% identity with a South American isolate (DQ915970), 99.7% homology with an Israeli isolate (U26740), and 99.4% identity with a Spanish isolate (AY394465). A phylogenetic tree of E. canis was inferred based on the 16S rDNA sequences obtained in this study and others (Fig. 2). Consistent with the results of previous studies [15,16,21], the E. canis 16S rDNA sequences generated in this study were very similar to those from other countries.
Nevertheless, there was substantial variation in the gp200 gene sequences between the Taiwanese samples and other previously reported isolates. According to the gp200 nucleotide sequences of the E. canis isolates from the United States (AF252298 and CP000107), the gp200 open reading frame was 4,266 bp in length and encoded a protein of 1,421 amino acids. The complete gp200 coding sequences derived from 17 Taiwanese samples were identical or nearly identical to each other (99.9~100% identities), even though the canine blood samples were obtained from different locations. The three representative sequences obtained from this study have been deposited in the GenBank database under accession numbers GU810148 (n = 8), HM067841 (n = 4) and HM067842 (n = 5). The only two nucleotide differences among these sequences occurred at positions 3591 and 3624. However, these variations did not cause any changes in amino acids. Notably, all of these sequences had three nucleotide insertions at positions between 4077 and 4078, 4081 and 4082, and 4082 and 4083 (based on the E. canis strain AF252298 numbering system) and thus consisted of 4,269 bp.
Comparative sequence analysis of the 4,269-bp products generated for the E. canis Taiwanese samples (TWN; provisionally designated as the TWN genotype) revealed that it had 97.9~98.0% homology with the corresponding sequences obtained from the USA Jake2 isolate (CP000107) and the Brazilian Sao Paulo isolate (EF636664), 97.8~97.9% homology with the sequence of the USA Jake1 isolate (AF252298), and 97.5~97.6% homology with the sequence of the Israeli 611 isolate (EF636665).
The deduced amino acid sequence of gp200 of the TWN genotype was aligned with the corresponding sequences of four other published isolates. When compared with the sequence of the USA Jake1 isolate, 79 amino acid changes scattered throughout the gp200 sequence of the TWN genotype were observed (Fig. 3). Of these changes, 15 amino acid residues occurred in two of five known species-specific antibody epitopes [14]. Overall, the TWN genotype had 94.4% homology with the amino acids of the USA Jake1 isolate, 94.6% homology with the USA Jake2 isolate, 94.5% homology with the Brazilian Sao Paulo isolate, and 93.7% homology with the Israeli 611 isolate.
A phylogenetic tree of E. canis was inferred based on the gp200 amino acid sequences obtained in this study and four other reported sequences [24]. In this tree, the gp200 sequence from the TWN genotype differed distinctly from other corresponding sequences of E. canis isolates from North America (the United States), South America (Brazil), and West Asia (Israel), and formed a separate phylogenetic clade (Fig. 4).
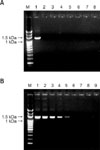
Finally, the specificity and detection limit of the four sets of PCR primers used for gp200 amplification (Fig. 5) were evaluated. For the purpose of specificity testing, the genomic DNA extracted from an E. canis-positive dog blood sample with 5% parasitemia was used to conduct PCR followed by agarose gel electrophoresis. The results showed that each of the four primer sets generated individual amplified products of the expected size, whereas no product was detected in reactions conducted without template DNA or using DNA that was separately extracted from blood samples of a healthy dog and dogs infected with different types of rickettsia, including Ehrlichia chaffeensis, Ehrlichia ewingii, Anaplasma platys, Babesia canis vogeli, and Babesia gibsoni. Among the four primer sets examined, EC200-F3/R3 exhibited the clearest pattern (Fig. 5A). Subsequently, the sensitivity of the PCR assay using this primer set was also determined. The blood sample with 5% parasitemia was serially diluted 10-fold with blood obtained from a healthy dog. The DNA extracted from these dilutions was used to determine the detection limit. As shown in Fig. 5B, the PCR results indicated that the lowest detection limit was 0.00005 parasitemia.
Discussion
While canine E. canis infection is considered to be enzootic throughout Taiwan, this conclusion is based on clinical signs, hematological abnormalities and microscopic examination of Giemsa-stained blood smears or serological evaluations such as enzyme-linked immunosorbent assays. In the present study, the nearly full-length 16S rDNA of E. canis from 17 infected dogs was amplified and sequenced, providing molecular evidence that this etiologic agent is involved in canine disease in Taiwan. Although the generated 16S rDNA sequences showed a very low diversity when compared with those from other geographically distinct isolates, additional analysis of the antigen-encoding gene gp200 helped identify genetic variations among E. canis isolates.
E. canis gp200 is a secreted nuclear translocated ankyrin repeat-containing protein that has five major species-specific epitopes that are primarily located in terminal acidic domains [14]. The protein has been shown to elicit strong antibody responses in the acute phase of the infection [24]. The development of a widely applicable vaccine for CME is undoubtedly dependent on an understanding of genetic differences that may exist in geographically dispersed strains of E. canis, particularly with respect to the genes coding for immunodominant antigens, such as gp200. Previous studies have reported that gp200 had a high level of conservation between the USA and Brazil isolates, but that substantial divergence was present in the Israeli isolate [14,24]. In the present study, we found that amino acid changes in gp200 from the TWN genotype were distributed throughout the protein, and that some of the amino acid substitutions occurred in known E. canis-specific epitopes [14]. Notably, most of the amino acid substitutions in epitope-containing regions were dimorphic, containing only two different amino acids at positions wherever substitutions occur. With respect to the Israeli isolate, it has been suggested that amino acid substitutions in gp200 epitopes are responsible for the lower immunoreactivity of the protein with heterologous antisera when compared with homologous Israeli antisera [24]. Accordingly, it would be interesting to determine if notable differences in the TWN genotype are involved in any antigenic variability.
Conversely, the sequence divergence in the gp200 gene among geographically distributed E. canis strains examined in this study and others has provided useful information regarding a possible new target for genotyping of the organism. The results presented here also expand our knowledge of the genetic variability of E. canis and encourage further research for the analysis of genetic variation of E. canis strains worldwide using additional samples. Further studies of E. canis isolates with greater global distribution may enable inference of the phylogeographic patterns of these strains. Considering that the clinical outcome of E. canis infection vary widely from death to an asymptomatic chronic carrier state in untreated animals [4,6,13,19], it would be of interest to assess the relationship between genetic variations in gp200 and strain infectivity or virulence of E. canis.
In summary, the present study documents infection of dogs with E. canis from Taiwan using molecular methods. Molecular evidence indicates that the E. canis genotypes circulating in dogs in Taiwan appear to be highly conserved. Based on phylogenetic analysis of the gp200 sequences, it is clear that the Taiwanese genotype reported here represents a novel strain of E. canis that has not yet been characterized. Additionally, the divergence of gp200 among geographically dispersed strains examined in this study and others supports the conclusion that this gene is useful for molecular genotyping of E. canis.




 XML Download
XML Download