

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Ovarian cancer is the most lethal cause of cancer-related death from gynecological malignancies in Western women [1,28]. Approximately 90% of ovarian cancer is believed to arise from a single layer of flat to cuboidal cells forming the outer layer of ovarian surface epithelium (OSE) [1,2,17,26] and OSE has received much attention in the last few years. However, the study of the etiology and cellular mechanisms of human ovarian cancer was limited because there has been no appropriate animal model to explain the occurrence of ovarian cancer. Although it has been reported that concurrent inactivation of Rb and p53 genes contributes to the initiation of ovarian cancer development [12], the cause of ovarian carcinogenesis is poorly understood. Brca1/2 and p53 are known to play important roles in DNA repair and the cell cycle. Recent evidence has shown that the mutation of the brca1 gene increases the incidence of preneoplastic changes in murine OSE and the inactivation of p53 reverses the increase of apoptosis induced by the loss of function of brca1 in vivo [9]. This indicates that concurrent inactivation of brca1 and p53 pathways may be involved in critical changes responsible for the induction of ovarian carcinogenesis.
Brca1 and brca2 are the DNA damage repair genes and are important in maintaining genomic integrity [7,30,32]. The mutation of these genes increases susceptibility to ovarian cancer [13] and the inactivation of brca1 induces the activation of a number of oncogenes involved in tumorigenesis [4,11,35]. The p53 protein is well known as a transcriptional factor and functions as a tumor suppressor gene. p53 also plays an important role in apoptosis and the cell cycle and its mutation is known to be involved in the incidence of 50% of human tumors [24]. Furthermore, there is accumulating evidence that the loss of brca1 induces the inactivation of p53, which is involved in brca1-associated tumorigenesis [5,10]. Although there have been many studies of the role of these genes in ovarian carcinogenesis, a definitive elucidation has not been achieved because an animal model of ovarian cancer in mouse OSE cells has not been established. The inactivation of brca1 induced developmental delay and resulted in embryonic lethality in mice [11,14,23]. Furthermore, deficiency of brca2 also induced embryonic lethality by the retardation of embryonic development [23,33]. To overcome these problems, the Cre-loxP system was employed to elucidate the roles of brca1 in tumorigenesis through the targeted deletion of genes. Using the Cre-loxP system, early studies showed that the conditional knockout of brca1 induces mammary tumor and also that concurrent inactivation of p53 and Rb1 is sufficient for the induction of ovarian carcinogenesis in conditional p53 and Rb1 knockout mice [12,36]. In this study, we investigated the role of brca1 or brca2 with p53 in ovarian carcinogenesis using the Cre-loxP system in floxed mice.
Materials and Methods
Cell culture
Ovaries were dissected and washed with phosphate-buffered saline (PBS; Invitrogen Life Technologies, USA). The ovaries were placed in DMEM/F12 medium containing Collagenase (100 U/mL) and Dispase II (0.6 U/mL) for 25 min at 37℃ in a humidified atmosphere. OSE cells were cultured in DMEM/F12 (Sigma-Aldrich, USA) supplemented with 10% FBS (Hyclone, USA), 100 U/mL penicillin G and 100 µg/mL streptomycin (Life Technologies, USA) at 37℃ in a humidified atmosphere of 5% CO2.
Mice maintenance
Homozygote mouse strains with brca1loxP/loxP, brca2loxP/loxP, and p53loxP/loxP were purchased from the Mouse Repository of Mouse Models of Human Cancers Consortium program (US National Cancer Institute, USA). FVB inbred mice (Charles River, USA) were used as a control group. The mice were maintained at a constant temperature (23 ± 2℃), relative humidity of 50~60%, and a 12 h light/dark cycle. Standard rodent chow and purified water were available ad libitum. All animal procedures were approved and carried out in accordance with the Animal Care Committee of the University of British Columbia.
Genotyping and DNA preparation
The mice carrying a brca1, brca2, and p53 conditional allele were genotyped by PCR (58℃, 1 min; 72℃, 1 min; 94℃, 1 min; 30 cycles). Total RNA extracts were prepared from the mouse cells using TRIZOL Reagent (Invitrogen Life Technologies, USA) and reverse transcribed from 2.5 ng of total RNA using the First-Strand cDNA Synthesis Kit (Amersham Pharmacia Biotech, USA). The sense/antisense primers for the floxed sequence and the wild-type sequence and the predicted sizes of PCR products of brca1, brca2, p53, and glyceraldehyde-3-phosphate dehydrogenase are described in Table 1.
Adenovirus administration
Mouse strains with wild type, brca1loxP/loxP, brca2loxP/loxP, and p53loxP/loxP were injected with adeno-Cre virus (AdCMVCre) and adeno-EGFP (AdCMVEGFP) (University of Iowa Gene Transfer Vector Core) as previously described [12]. Briefly, the adenovirus was injected using a 1 mL syringe with a 30-gauge beveled needle after 0.5~2% isofurane anesthesia. Ovaries were individually accessed via dorsal incision and the right ovaries were injected with AdCMVCre and the left ovaries were injected with AdCMVEGFP.
For in vitro infection, 2 × 105 OSE cells were incubated with 4 × 107 AdCMVCre, and AdCMVEGFP (MOI 200, respectively) in 500 µL of serum-free medium for 1.5 h at 37℃. Then the cells were washed twice with PBS and incubated in DMEM/F12 with 10% FBS. Cells isolated from brca1loxP/loxP and brca2loxP/loxP mice were infected with AdCMVCre to conditionally delete brca1 and brca2 genes. A multiplicity of infection (MOI) value was calculated as the number of infectious viruses divided by the number of cells.
Proliferation assay
The proliferation assay was performed using [3H]-thymidine incorporation assay as previously described [8]. Briefly, cells were placed in 24-well plates with 0.5 mL DMEM/F12 with 10% FBS. The cells were infected with the adenovirus as described above. Following incubation for 2 days, 1 µCi [3H]-thymidine (0.5 Ci/mmol; Amersham Pharmacia Biotech, USA) was added. The cells were incubated for 6 h and precipitated with 0.5 mL 10% trichloroacetic acid for 20 min at 4℃. The precipitate was washed in methanol twice and solubilized in 0.5 mL 0.1 N sodium hydroxide. The radioactivity was measured in the Tri-Carb Liquid Scintillation Analyzer (Model 2100TR; Packard Instrument, USA).
Histopathological analysis
Brca1, brca2, brca1/2, and brca2/p53 floxed mice were injected intrabursally with AdCMVCre and AdCMVEGFP into the right and left ovaries, respectively. brca1loxP/loxP, brca2loxP/loxP, or p53loxP/loxP mice infected with AdCMVCre and AdCMVEGFP adenovirus were sacrificed by carbon dioxide inhalation according to the standard protocol. Ovaries were removed and fixed in 10% formalin, and paraffin sections were prepared for microscopic evaluation. Serial sections were prepared for hematoxylin and eosin staining, and histopathological examination then performed under a light microscope.
Results
Detection of the insertion of loxP sites in brca1 and brca2
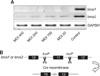
The insertions of loxP sites in mice carrying a brca1, brca2, and p53 conditional allele were confirmed by RT-PCR. As demonstrated in Fig. 1A, predicted PCR products of 5' floxed brca1 and 3' floxed brca1 were obtained at 461 and 562-bp, respectively, and 5' and 3' floxed brca2 PCR products were observed at 376 and 529-bp, respectively (Fig. 1B). PCR products from the wild type of brca1 and brca2 mice were obtained with the same primer sequences of brca1 and brca2. The mice bearing floxed p53 gene as confirmed by RT-PCR were used for further experiments (Fig. 1C. Lanes 3 and 5).
Determination of MOI to delete the loxP sites of brca1 and brca2 following AdCMVCre infection
Cre-loxP recombination was performed to inactivate brca1 and brca2 genes. For experiments, 3 × 105 cells were seeded in 0.5 mL of serum-free medium with serial dilution of AdCMVCre. After 1.5 h incubation at 37℃, the cells were washed twice with PBS and covered with DMEM/F12 containing 10% FBS. Infection of AdCMVCre at MOI 20~400 determined by the formula resulted in a decrease of brca1 and brca2 expression (Fig. 2A), indicating that AdCMVCre administration activated the loxP sequence, which successfully excised the brca1 and brca2 genes as described in a schematic diagram (Fig. 2B).
Effect of the inactivation of brca1, brca2, and p53 and on the proliferation of OSE
OSE cells were obtained from brca1loxP/loxP, p53loxP/loxP, brca1/2loxP/loxP, and brca2/p53loxP/loxP mice and infected with AdCMVCre at MOI 200 to investigate whether the inactivation of brca1, brca2, and p53 affected the proliferation of mouse OSE cells. Data from the infection group of AdCMVCre were compared to those of the vehicle group or the AdCMVEGFP infected group. Infection with AdCMVCre in OSE cells resulted in a significant increase of proliferation in brca1loxP/loxP and brca2/p53loxP/loxP groups (Fig. 3), but no difference was observed in brca1/brca2 floxed OSE cells (Fig. 3). Although the infection of p53loxP/loxP cells with AdCMVCre seemed to increase proliferation of OSE, the increase was not significant (Fig. 3).
Effect of the inactivation of brca1 and brca2 on ovarian carcinogenesis
To evaluate the effect of the deletion of brca1 and brca2 genes, wild type, brca1loxP/loxP and brca2loxP/loxP mice were injected with AdCMVCre intrabursally. Following 3 months of AdCMVCre infection, ovaries were collected and examined under a light microscope following staining. No morphological changes were observed in the OSE cells of brca1 and brca2 floxed mice (data not shown).
Since the effect of concurrent inactivation of brca1 and brca2 in ovarian carcinogenesis is not known, double knockout mice of these genes were obtained by cross breeding. There were no observable differences between AdCMVCre and AdCMVEGFP infected groups. In addition, no morphological changes were observed in brca2/p53loxP/loxP mice following AdCMVCre administration (data not shown).
Discussion
The carriers of brca1 and brca2 mutations have increased susceptibility to breast, prostate, and ovarian cancer [13,35]. The unique signatures of gene expression profiling of brca1 and brca2 have been revealed in hereditary and sporadic ovarian cancers [16]. In addition, a significantly higher proportion of advanced serous adenocarcinoma in ovaries has been observed in the cases of brca1 and brca2 mutations [31], indicating that the loss of function of these genes leads to the outgrowth of ovarian cells. Somatic mutations also seem to be involved in ovarian cancer development [6,19]. The germline mutations of brca1 and brca2 genes in ovarian carcinomas demonstrate a higher growth percentage than sporadic cancers do [20,22]. It has been reported that brca1 dysfunction is often involved in simultaneous brca2 dysfunction in ovarian cancers [18]. Although many studies have been performed to elucidate the precise role of brca1 and brca2 on ovarian carcinogenesis, the studies have struggled to define the exact contribution because the knockout of these genes in mice induced developmental delay and resulted in embryonic lethality [11,14,23,33].
Previous study has shown that when using a conditional knockout method, the Cre-loxP system, mutation of brca1 resulted in mammary tumor formation in the mouse [36]. Also, a single intrabursal administration of recombinant Cre-expressing adenovirus in double-floxed mice of p53 and Rb1 was sufficient for reproducible induction of ovarian epithelial carcinogenesis [12]. In this study, we investigated whether the concurrent mutation of brca1, brca2, and p53 genes is involved in ovarian carcinogenesis. Following the confirmation of genomic phenotype of floxed brca1, brca2 and p53 in brca1loxP/loxP, brca2loxP/loxP and p53loxP/loxP mice, double homozygous mice of brca1/2loxP/loxP or brca2/p53loxP/loxP were obtained by cross breeding of the single-floxed mice with brca1loxP/loxP, brca2loxP/loxP, and p53loxP/loxP mice. We confirmed the loss of copies of brca1/2 and brca2/p53 in brca1/2loxP/loxP or brca2/p53loxP/lox mice by genotyping using PCR. To explore the function of brca1, brca2, and p53 in OSE in vivo, AdCMVCre was injected intrabursally to induce Cre-loxP recombination to delete the exons of brca1 and brca2. We examined tumor formation after 3 months of AdCMVCre infection. No morphological changes were observed in AdCMVCre injected ovaries. A previous report has shown that the inactivation of brca1 is sufficient for preneoplastic changes such as hyperplasia and inclusion cysts following adeno-viral Cre infection but no tumor formation was observed [9]. There is also controversy over the issue of the effect of inactivation of brca1 on the capacity of proliferation of OSE cells [3,9,29]. Discrepancies might be due to the different viral constructs or the period of infection. In addition, no susceptibility to mammary tumors was observed in heterozygous brca1 mutant mice [37], suggesting that the existence of the normal functioning brca1 gene is possible.
P53 is well known to be absent or mutated in several cancer types and the dysfunction of p53 is commonly related to ovarian cancer, particularly with germline brca1 muation [21,27]. brca1/2 are known to have similar phenotypic consequences of disruptions [34]. Since the precise nature of brca1/2 and p53 interaction remains to be defined, we evaluated the effect of the mutation of these genes on OSE proliferation. OSE cells obtained from the ovaries of conditional knockout mice were infected with AdCMVCre to delete the exons of brca1, brca2, and p53 in vitro. Infection with AdCMVCre in brca1loxP/loxP, brca2/p53loxP/loxP OSE cells resulted in an increase of cell proliferation, but no proliferative increase was found in the AdCMVCre infected brca1/2loxP/loxP group and p53loxP/loxP group. In addition, no morphological changes were observed in brca2/p53loxP/loxP mice following AdCMVCre administration. However, it has been reported that the individual inactivation of p53 and Rb1 increases OSE proliferation in vivo and the concurrent inactivation of those genes demonstrates a synergistic effect on cell proliferation [12]. These discrepancies may be partially due to the experimental methods used to measure the proliferative index, since the level of proliferation is similar among these results. However, our results are consistent with the finding that the concurrent mutation of these genes was not necessary in breast cancer development [15]. In addition, a previous report showed that brca2 interacts with p53 gene and inhibits p53's transcriptional activity in cancer cells [25], suggesting that the concurrent inactivation of these genes may not be involved in ovarian cancer development and that it is more likely that other genes may play a role in ovarian carcinogenesis. It is possible that a single intrabursal administration of AdCMVCre was not sufficient to inactivate these genes and induce ovarian cancer in vivo and further studies using more frequent administration or long term maintenance following AdCMVCre infection are needed before a conclusion can be reached.
In conclusion, we demonstrated in this study that the proliferation of OSE was significantly increased in mice bearing concurrent floxed copies of brca1 and brca2/p53 in vitro. However, we did not observe direct evidence of the involvement of brca1, brca2, and p53 in ovarian carcinogenesis. Also, morphological changes including tumor formation were not observed in mice having floxed copies of concurrent brca1/brca2 or p53 following an intrabursal administration of recombinant Cre-expressing adenovirus in vivo. Taken together, these results suggest that the inactivation of brca1, brca2 or p53 is unlikely to be sufficient for the induction of ovarian epithelial carcinogenesis.



 XML Download
XML Download