

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Foot-and-mouth disease (FMD) is a highly contagious viral disease of cloven-hoofed animals, both domestic and wild [8,18]. The disease is caused by FMD virus (FMDV), which is one of two species in the genus Aphthovirus in the family of Picornaviridae, an important group of mammalian RNA viruses [3,11]. FMD has been endemic in many continents, including Africa, South America and Asia. It has had a dramatic impact on the farming industry leading to tremendous economic losses particularly in countries which are naturally FMD-free [20]. Previous studies have shown that Iran has one of the highest reported rates of FMD cases per year. The high incidence of FMD (including serotypes A, Asia 1 and O) has allowed the identification of new variants of the virus over the last 7 years. [16]. Furthermore, due to high error rates in the process of genome replication, FMDV is genetically highly variable. It has been grouped into seven serological serotypes (O, A, C, Asia 1, SAT 1, SAT 2 and SAT 3) and multiple subtypes, with more than 60 subtypes of the virus now characterized [13]. This large antigenic variation is considered one of the major obstacles for the control of FMD by vaccination in endemic areas [1,4,9]. Non-enveloped FMDV contains a single-stranded, positive-sense RNA genome with approximately 8,500 nucleotides. The open reading frame (ORF) encodes a single polyprotein which can be cleaved into four structural proteins (VP4, VP2, VP3, and VP1) and eight non-structural proteins (L, 2A, 2B, 2C, 3A, 3B, 3C, and 3D) [6,17]. In general, structural proteins are more variable than non-structural proteins. Mutations or deletions in structural proteins may help FMDV to evade an immune response produced by the host, whereas mutations or deletions in the non-structural proteins could be detrimental to viral replication and protein processing [7]. In the non-structural protein regions, the 2A, 2B, 2C, 3C, and 3D proteins are highly conserved, while the 3A and 3B proteins are less conserved among serotypes, probably due to their functions or actions with host factors [12]. The wide variation of serotypes across different geographical areas points to the need for urgent identification of regional serotypes for outbreak control and vaccine development.
The purpose of this study was to determine the full-length nucleotide sequence of genes encoding 3A and 3B proteins of A Iran 05 subtype, and to compare it with available corresponding sequences deposited in the GenBank database.
Materials and Methods
Viruses
In 2008, clinical specimens of FMDV which included tongue epithelium tissue were collected from one of the Iranian fields located in Tehran. They were sent immediately to the laboratory of Razi Vaccine and Serum Research Institute in Karaj. Laboratory and serological tests for FMDV confirmed the presence of infected samples. The new virulent isolate was passaged in baby hamster kidney cell monolayer cultures, and the infected cell culture supernatant was clarified and stored at -70℃ before use.
RNA extraction, reverse transcription-polymerase chain reaction (RT-PCR) and sequencing
The infected cell culture supernatants were used for the extraction of viral RNA using the Total RNA Isolation kit (Roche, Swiss) following the recommendations of the suppliers. The 3AB coding region of FMDV is 672 nucleotides long and was amplified by standard methods in a one-step RT-PCR, using the primer combination forward G10 (5'CCAAGGA CGGGTACAAAGTTAAC3') and reverse G11 (5'ACCAT CTTTTGCAAGTCG/AGTC3'). The amplified PCR products (672 bp) of the expected length were subjected to electrophoresis in a 1% agarose gel and visualized by staining with ethidium bromide under UV transilluminator. After the successful amplification of the target DNA sequence, fragments were purified using gel extraction kit (Roche, Swiss) in accordance with the manufacturer's instructions. Purified PCR product was cloned into PNTZ57T vector (Fermentas, Germany) according to the manufacturer's instructions and sequencing was carried out using T7 promoter primer (MWG Biotech, Germany).
Sequence analysis and phylogenetic tree construction
A fragment of 672 nucleotides containing the full length 3A and 3B regions (edited manually) enabled us to perform multiple sequence alignments using the BioEdit software, version 7 [10]. To determine the degree to which genetic diversity is reflected in the 3A and 3B proteins, alignment and comparison of the deduced amino acid sequences of isolates were carried out (Fig. 1). The published sequences of 20 FMDV type A isolates recovered from different parts of the world were included in this analysis and compared with the corresponding sequence of A Iran 05 isolate. The phylogenetic tree was constructed using the Neighbour Joining method in the program Clustal X, version 1.8 [19]. The nucleotide sequence homology/divergence was calculated using the MegAlign project of DNAStar software package version 5.1 (DNAStar, USA). A description of type A FMDV isolates has been given in Table 1.
Results
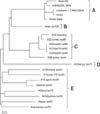
The sequences for the non-structural protein coding regions (3A and 3B) were determined by PCR amplification and sequencing. A nucleotide sequence comparison based upon the alignment of complete nucleotide sequence of the 3AB region indicated that A Iran 05 had the greatest sequence similarity with four isolates (AIRN2005_WRL, Lindholm 1.3 PAK32006, PAK1, and PAK5 2006) with a nucleotide identity of approximately 98% (data not shown). Fig. 2 shows a phylogenetic tree constructed based on the sequence alignment of 21 genomes, which are distinctly divided into five lineages. The isolate A Iran 05 clustered with a Turkish and three Pakistan isolates into a separate branch from other type A isolates (lineage A). The A Iran 05 isolate was closely matched with viruses isolated in Turkey and Pakistan during 2005~2006. All the field isolates (except for isolates of lineage A) shared comparatively lesser homology (81~89%) with the Iranian isolate (A Iran 05). The topology of the phylogenetic tree indicated that the isolates airan iso105 and a21kenya iso77 differed from the Iranian subtype and could be placed in the lineages B and D, respectively (Fig. 2). All a22 type A isolates and lone isolate a28turkey iso44 were grouped closely in lineage C (Fig. 2).
The remaining eight isolates of the present study originated from different geographical areas and formed a single group (lineage E). A minimum homology of 81% in nucleotide sequence was noted between the A Iran 05 isolate and the Kenyan isolate a21kenya iso77. Analysis of sequence data revealed that the Iranian isolate (airan iso105) described earlier has a low similarity to A Iran 05 with a sequence identity of 89%. Comparison of amino acid sequences identified amino acid changes in both the non-structural proteins. The number of sequence differences exhibited by each of the isolates revealed that A Iran 05 contains two amino acid substitutions at positions 47 and 119 of the 3A coding region (Fig. 1). Comparison of 3B protein among the different variants showed that in relation to the A Iran 05 the entire isolates showed a change at position 27. Apart from this, no variation leading to amino acid substitution was found in 3B protein. In 3A protein, the N-terminus (1~33) amino acids were highly conserved (Fig. 1A) while the C-terminus amino acids were highly variable. In comparison to the 3A protein, the N-terminus (1~17) amino acids of the 3B protein were highly variable (Fig. 1B). In general, the 3A and 3B coding regions were prone to amino acid alterations. As can be visualized in Fig. 1, there is obvious evidence of amino acid substitutions in lineage A, during 2005 from Iran up to the 2006 from Turkey and Pakistan. From the results highlighted in Fig. 1, it becomes evident that in relation to the A Iran 05, four other isolates of lineage A had changes at residues 3A (Gly17→Ala, Ser135→Gly and Asn139→Glu) and 3B (Leu53→Pro).
Discussion
Analysis of the viral genome sequence is of importance to monitor the field isolates in areas where the disease is endemic. Continuous co-circulation of FMDV in the field and lack of proofreading activity of the viral polymerase in the replication process results in the emergence of many genetic variants [2,5]. Within the FMDV serotypes, serotype A displays the greatest number of newly occurring subtypes [13]. The Middle East has been severely affected by separate type A (A Iran 05) epidemics, which initially emerged in Iran in 2005 and moved westwards into Turkey (including the European part of Thrace). It has continued to spread in 2006, circulating in Turkey and Iran, and has been also detected in Pakistan, Saudi Arabia and in the first months of 2009 in Iraq, Kuwait and Bahrain, and was recently identified in samples sent by Lebanon and Libya to the World Reference Laboratory [18]. This particular FMDV subtype has proven to be highly virulent and has caused severe disease in all ages of cattle. Variation in virulence could be due to the quasispecies structure of FMDV populations and the error-prone replication process. Together with this mechanism, recombination within the non-structural genome regions, potentially modifying the virulence of the virus, may be involved in the success of this subtype [13]. Previous studies have shown that both regions (3A and 3B) appear to influence the virus's pathogenic potential [14].
It is thought that virus circulation is limited to some areas of western Asia and southern Asia. Iran is bordered on the east by Pakistan and on the west by Turkey and Iraq. Permeable borders and live animal trade in west Asia are likely reasons for virus circulation. Throughout the period of virus circulation, amino acid substitutions accumulated at the various sites of protein sequence. The genetic polymorphism amongst different subtypes of type A demonstrated that in the 3A coding region of isolate A Iran 05, two nucleotides 139 and 355 were changed from T to C and G to A, respectively, resulting in exchange in amino acids from Phe47→Leu and Ala119→Thr. In addition, there is a substitution at nucleotide 79 (G to A) of 3B region, which leads to an amino acid exchange of Ala27→Thr. Surprisingly, in contrast to A Iran 05 isolate the remaining twenty isolates showed no corresponding changes in the mentioned positions. This trend leads us to suggest that evolutionary events are occurring. The low conservation of the non-structural protein 3A could be due to its putative role in the adaptability of the virus to the host [12]. The phylogenetic analysis based on the 3A and 3B genes is in complete agreement with a previous report documenting that four isolates of lineage A have significant sequence similarity [13]. Nucleotide sequence analysis of the field isolates showed that virus circulation during 2005~2006 in Iran, Pakistan and Turkey may have originated in Iran. Analysis of the amino acid sequences revealed that five isolates of lineage A had four conserved regions (Leu114→Gln), (Ser117→Asn), (Gly118→Ser) and (Ala143→Thr) in the 3A protein. The other viruses revealed no such changes. Therefore, these residues might be looked upon as signature amino acids for the group of lineage A field isolates.


 XML Download
XML Download