

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
Cryptosporidium (C.) spp. are members of the phylum Apicomplexa and found in human and animal populations worldwide. People from both developed and developing countries are vulnerable to these opportunistic protozoa. It has a predilection for epithelial cells in the digestive tracts of a wide variety of hosts, including humans, livestock, companion animals, wildlife, birds, reptiles and fishes [16]. This protozoan is responsible for moderate to severe opportunistic infection in both immunocompetent and immunocompromised individuals, the latter group being more susceptible with potentially fatal consequences. The immunocompetent individuals usually experience a self-limiting disease often manifested by acute profuse, watery diarrhoea accompanied by abdominal pain and other enteric symptoms like vomiting, low grade fever, general malaise, weakness, fatigue, loss of appetite, nausea, chills and sweats. Furthermore, the disease may be chronic and even life threatening for undernourished infants and AIDS patients [12].
Mass spectrometry based BLAST (MS BLAST) is a database search protocol for identifying unknown proteins by sequence similarity to homologous proteins using peptide sequences produced by mass spectrometry [5]. It also can utilize redundant, degenerate, and partially inaccurate peptide sequence data derived from de novo interpretation of MS/MS spectra. The use of MS BLAST and its efficiency and limitations has been reviewed by Habermann et al. [5]. Similar attempts using high scoring pairs (HSPs) have been described by other authors [22,24] where protein characterisations were performed by exploitation of the genome sequence data. As the ungapped BLAST identifies all HSPs between individual peptides in the query, the sequential order of the matched segments does not influence the total score (which is calculated for each protein hit by adding up the scores of individual HSPs that are higher than the threshold).
Identifying the proteins of any organism with an incomplete genome sequence is also possible with MS BLAST. Shevchenko et al. [22] proposed that identifying proteins from the yeast Pichia pastoris, for which the whole genome sequence was not available at that time, was possible using MS BLAST approach. However, they used a different submission technique to query sequences for BLAST searching. All complete and partial peptide sequences obtained from MS data interpretation were edited before the BLAST search, where the sequence of peptides were spaced with the minus (-) symbol and were merged into a single string. They proposed that the gap symbol (-) assigns a high negative score in an algorithm which prevents false similarities to the sub-sequences (including parts of peptide sequences adjacent in a query string).
The comparative efficiency of MS-Shotgun, FASTS and MS BLAST on a small dataset of peptide sequences from 14 proteins of the 20S proteasome of Trypanosome brucei indicated a similar efficiency among these three protocols [5,11]. In another study, MS BLAST was found to double the number of microtubule-associated proteins from the African clawed frog Xenopus laevis compared with conventional database searching [10]. However, information regarding the peptide (minimum length, percent identity and number of fragmented peptides) sufficient for identifying homologous proteins (in another species) is yet not established. The cross species identification of proteins by MS BLAST protocol has been evaluated using computer modelling, where it was found to be promising and useful like FASTS and FASTF [5]. The study also showed that within the mammalian subkingdom, over 80% of proteins could be positively identified by sequence similarity searches.
Recently the partial proteome of C. parvum sporozoite has been reported with 30% coverage of the total predicted proteome [20]. Their lies the need for complementary approaches to further characterize the remaining proteome for any comprehensive analysis. The aim of this study was to employ the bioinformatic tools to analyse the proteome of the sporozoite stage of C. parvum using the MS data from the 1D-SDS-PAGE with LC-MS/MS analysis and a separate multi-dimensional protein identification technology (MudPIT) analysis of whole sporozoite lysate. Alongside the use of MASCOT search software for analysis of MS data, the MS BLAST search protocol has been used to optimize the use of peptide sequence information derived after MS analyses.
Materials and Methods
Chemicals and oocyst materials
All chemicals were purchased from VWR (UK) unless otherwise specified. DTT, CHCA, iodoacetamide, and EDTA were obtained from Sigma Aldrich (UK). Modified porcine trypsin was a product of Promega (UK). HPLC grade acetonitrile, HPLC grade methanol and glacial acetic acid was purchased from Fisher Scientific (UK). Oocysts of C. parvum passaged in lambs (IOWA strain) were purchased from Moredun Research Institute (MRI, Scotland). This strain was continually passaged in sheep by MRI. Oocysts were concentrated by sucrose density centrifugation, washed and resuspended in phosphate-buffered saline (PBS; pH7.2). The parasite suspension was stored at 4℃ in the presence of 1,000 U per mL penicillin and 1,000 µg per mL streptomycin.
One dimensional SDS-PAGE
For one dimension electrophoresis, frozen sporozoite pellets were disrupted in 40 µL of gel loading buffer containing 50 mM Tris Hydrochloride (pH 6.8), 100 mM DTT, 2% (w/v) SDS, 0.1% (w/v) Bromophenol blue and 10% glycerol. The mixture was boiled at 100℃ for 10 min and chilled on ice before loading into the SDS-PAGE gel lane. A standard broad-range protein molecular weight marker (RPN 5800; Amersham Biosciences, UK) was used as the ladder in a separate lane. Polyacrylamide gels (12%) were made using a mini gel apparatus (BioRad, UK). The resolving gel consisted of 30% acrylamide in 1.5 M Tris-HCl (pH 8.8), 10% (w/v) SDS, 10% (w/v) ammonium persulphate (APS) and 10 µL N,N,N',N'- tetramethylethylenediamine (TEMED). The stacking gel consisting of 30% acrylamide in 1.5 M Tris-HCl (pH 6.8), 10% (w/v) SDS, 10% (w/v) APS and 5 µL TEMED was used for the quantification of protein extracts. The SDS electrophoresis buffer was prepared by dissolving 25 mM Tris-base, 192 mM glycine and 0.1% (w/v) SDS in 400 mL of double distilled deionised water. Separation was performed by electrophoresis at 120 V for 2 h and then the gels were stained with Coomassie Brilliant blue or by Colloidal coomassie staining technique [15].
MudPIT analysis
Two-dimensional-nLC-MSMS analysis was performed using an Ultimate 2D nLC system (Ultimate Famos Switchos; Dionex, USA) in the standard configuration, interfaced via a 20 µm i.d 8 µm orifice Picotip (New Objective, USA) mounted on a Protana nanospray interface (Protana, Denmark) to a QStar Pulsar i mass spectrometer running the AnalystQS software (Applied Biosystems, USA). A 1 × 15 mm BioSCX trap, 0.3 × 5 mm PepMap trap and 75 µm × 15 cm PepMap column were used in the analysis (Dionex, USA). Flow rates were 30 µL min-1 on the high flow side and approximately 200 nL min-1 on 93 the low flow side. 10 salt cuts at 0, 20, 40, 60, 80, 100, 150, 200, 300 and 500 mM KCl were used and a gradient of 2~50% acetonitrile in water with 0.5% formic acid for the reversed phase separation. Data was collected using an IDA protocol with a 2s survey scan 400~2,000 Da, and the four most intense ions above a threshold of 20 counts not on the exclusion list chosen for analysis using 3s MSMS scans in the 50~2,000 Da range. Masses were then added to an exclusion list for 360s.
The Cryptosporidium database
The CryptoDB proteome database (release 3.1) was used as a source to download the genome, EST and GSS datasets into a local server connected to the mass spectrometer.
NCBI and other protein databases
The MASCOT searching of MS data was performed either against the non-redundant National Centre for Biotechnology Information (NCBI, USA) database or locally downloaded CryptoDB datasets.
The MASCOT search tool
The MASCOT search engine (Matrix Science, USA) was used to analyse the PMF and peptide fragmentation data. The MASCOT search against the genome sequence of C. parvum revealed a list of contigs with significant scores for individual peptides. The ion scores of the individual peptides were recorded from the MASCOT search output page and the BLAST searching of any putative protein sequence was performed through the linked web from the same page. However, this was not suitable in cases where the significant peptides were few in number, or located some distance apart in a long contig.
BLAST and MS BLAST
The MASCOT search against the NCBI database and locally downloaded Cryptosporidium genome sequences revealed a list of contigs with significant peptides. The sequence containing those peptides was then BLAST searched (protein-protein BLAST or BLASTp) to identify sequence similarity with proteins from other organisms. The interpretation of the score and sequence similarity from BLAST searching eventually led to the identification of putative or homologous protein sequences. The whole sequential steps of this data analysis towards the identification of putative or homologous sequences are illustrated in Fig. 1.
Briefly, the PMF data and peptide fragmentation data from MS analysis was searched against the NCBI database and a number of contig hits were revealed, for which it matched one or more peptides with a specific ion score for each of them. Once the Mascot score was found significant (as manifested by a direct match with a protein or EST in the database with a significantly high individual peptide score for which the entry was already submitted in the database) the identity was confirmed for that protein or its homologs. However, if the MASCOT score was not significant and the identified peptides had a high ion score or if they were closely located together (indicating peptides from one protein), they were further searched against the CryptoDB database. The search again revealed some contig hits and the relevant peptides, with or without a significant MASCOT score. The peptides with insignificant ion scores were then discarded while those with high MASCOT scores were used for further MS BLAST analysis.
Identification of proteins by MS-BLAST search
The MS BLAST strategy involved the use of BLAST search tools and the putative protein sequence containing the significant peptides identified by mass spectrometry. The sequence string was carefully chosen for the MS BLAST approach. Usually, two or more peptides which were located closely enough to be a part of a single protein were submitted for BLASTp search. This was done by submission of the sequence string from the beginning of the first peptide until the end of the last peptide for a BLASTp homology search. The output of the BLASTp search was then further analysed to identify putative protein hits. The length of the query sequence was recorded for each search. Once the significant hits were identified, the number of search peptides (in the query sequence), the GenBank accession number of homologous sequences, names of the proteins, the percentage of sequence similarity and the position of the query sequence in the contig were recorded.
Functional cataloguing of identified proteins
The gene ontology (GO) analysis provides valuable information to assign a putative function for any identified protein [8]. The three general principles of GO were molecular function, biological process and cellular component. As the gene product had one or more molecular functions and was used in one or more biological processes, it is likely to fall into subcategories for one or more of these broad ontology groupings. Using the GO databases (AmiGO, USA), the GO number was checked for any protein or its homolog in other species.
MIPS functional catalogue database (FunCat DB)
The FunCatDB (MIPS, Germany) is an annotation scheme for the functional description of proteins from different prokaryotes, unicellular eukaryotes, plants and animals. It consists of 28 main functional categories, including different functional categories such as cellular transport, metabolism, cellular communication/signal transduction, etc.
Bioinformatics-Harvester (EMBL) database
The Bioinformatic Harvester EMBL Heidelberg [9] is a protein database which collects and displays bioinformatic data and predictions for human proteins from various databases. The database collects text-based information from the a number of public databases and prediction servers which includes Uniprot, SOURCE, Genome Browser, BLAST, SMART, SOSUI, PSORT II, CDART, MapView, NCBI-BLAST, SOSUI, STRING, Genome Browser and EMBL. Once the data are downloaded and saved, it is subsequently presented as text or inframe, depending on the data presentation of the original server. Therefore, it provides similar result as in the original database. For this experiment, the gene ontology number and related information of any significant entries (from BLAST searching) were derived from the reference proteome published in this database.
Results
Identification of C. parvum proteins by MS-BLAST searching of 1D-SDS-PAGE data
The MASCOT search of LC-MS/MS data (while searched against the non redundant NCBI database) from all 20 samples in 1D-SDS-PAGE gel bands (Fig. 2) revealed 33 hits of Cryptosporidium. To obtain further information from the same MS data, the MS BLAST strategy was applied for a sequence similarity based protein homology search. While the mass fragmentation data of each individual band from LC-MS/MS were searched against the locally downloaded Cryptosporidium ORF (open reading frame) dataset, a total of 196 significant hits against contigs were recorded from 20 searches. When the contig sequences were analysed, each contig was found to contain at least one significant peptide hit, while some contained as many as 20 significant peptides hits (data not shown). In many instances the identified peptides with significant scores were found closely situated in a long continuous contig. The predicted ORF sequences (with significant peptides within each sequence string) were then used for BLAST search (protein-protein BLAST) for homology based protein identification. A total of 165 Cryptosporidium proteins were identified by this MS-BLAST approach. However, those hits included both C. parvum (n = 84) and C. hominis (n = 81) entries. In nearly all cases, the C. hominis homologous proteins were found with the same query in MS BLAST and the peptides were almost identical to C. parvum. Incorporating the two protein lists from the 1D-SDS-PAGE experiment (derived by MASCOT searching against the non redundant NCBI database and a MS BLAST search with peptides from Cryptosporidium ORF dataset) identified 100 C. parvum proteins (Table 1). Comparing the two approaches, the MS BLAST search strategy was found to provide 5 times greater (33 to 165) information than the NCBI search alone.
Many hypothetical proteins (n = 37) were identified by bioinformatic analysis of 1D-SDS-PAGE experimental data and the high MASCOT score along with higher percent identity confirms their physical existence in the proteome. Again, a number of metabolic enzymes have been identified, which include protein disulphide isomerase (gi.32398654), glyceraldehyde-3-phosphate dehydrogenase (gi.46229140), phosphoenolpyruvate carboxylase (gi.46227248), phosphoglucomutase (gi.46227774), glucose methanol choline oxidoreductase, enolase (gi.46227284), fructose, 1,6 biphosphate aldolase (gi.46227620), pyruvate kinase (gi.46227634) and phosphoglycerate kinase (gi.46229859). Several membrane associated proteins (gi.32398735, gi.46228663, gi.46227005) and oocyst wall protein (gi.46226838) were also identified. Other groups of proteins include many ribosomal proteins (n = 24), heat shock proteins (gi.2894792, gi.17385076, gi.46229711), and several uncharacterised proteins with unknown functions.
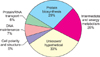
The functional categorization of 84 identified C. parvum proteins from the Cryptosporidium ORF dataset were made according to MIPS functional catalogue database (Fig. 3). The protein hits were matched with the human protein database, with the GO number and relevant functions of the homologous protein hits recorded for further analysis. A third (33%) of the identified proteins constituted hypothetical proteins while another third (29%) were responsible for protein biosynthesis. A significant proportion (20%) of total hits were proteins involved in intermediate and energy metabolism, while other groups were involved in DNA maintenance (7%), protein/RNA transport (6%) and proteins responsible for cell polarity and structure (5%).
Identification of C. parvum proteins by MS-BLAST searching of MudPIT data
The MudPIT analysis of sporozoite protein revealed a total of 42 proteins of Cryptosporidium while searched against NCBI database (Table 1). However, the number of submitted Cryptosporidium entries (i.e. previously identified and characterized) in NCBI is limited which possibly limits the success of such analysis. Therefore, the MS BLAST strategy was applied for sequence similarity based protein homology searching from the peptide fragmentation data derived after MudPIT analysis. While the MudPIT data were searched against the locally downloaded Cryptosporidium ORF dataset, a total of 150 hits of significant contigs were recorded. As previously observed in 1D-SDS and MS BLAST, the number of significant peptides in each contig also ranged from 1 to 20. The Cryptosporidium ORF sequence strings containing those significant peptide(s) were then used as a query sequence for BLASTp homology searching. The protein-protein BLAST searching revealed a total of 259 proteins of Cryptosporidium sp. which included a wide range of proteins. However, they included proteins from both C. parvum (n = 133) and C. hominis (n = 126). As with the MS BLAST analysis following 1D-SDS-PAGE, the homologous proteins of C. hominis were found in the same query for MS BLAST where the peptides were identical as in C. parvum. Notably, a similar level of redundancy was observed between C. hominis and C. parvum proteins. Incorporating the two protein lists from the MudPIT experimental data analyses provides a total of 140 proteins of C. parvum. Comparing the two approaches, the MS BLAST search strategy was found to be more informative in that it provided 6 times higher information than MASCOT search alone (42 to 259). A number of hypothetical proteins (n = 17) were identified by MS-BLAST search while many metabolic enzymes were recorded during the analysis. Some of the important enzymes are protein disulphide isomerase (gi. 32398654), enolase (gi.46227284), alcohol dehydrogenase (gi. 46228815), glycogen phosphorylase (gi.46229042), lactate dehydrogenase (gi.46229853), glyceraldehyde-3-phosphate dehydrogenase (gi.46229140), fructose,1,6 biphosphate aldolase (gi.46227620), pyruvate kinase (gi.46227634), NADP+ oxidoreductase (gi.13897519), phosphoglycerate kinase (gi.46229859). Several oocyst wall proteins and mucin like surface glycoproteins were also revealed from the study. In addition, a quarter of the total proteins (n = 35) identified from MudPIT analysis consists of 40S and 60S ribosomal proteins.
Discussion
An issue concerning MS BLAST is the quality of the spectra generated by MS and whether the software could efficiently analyse the spectra to detect the correct region of peptide sequence [22]. Again, as different MS analyses produce different patterns of peptides, MASCOT and MS BLAST could be combined as an integrated search tool. To optimise the use of peptide information, an alternative approach of MS BLAST searches proved useful in this study. This strategy enabled up to 6 times higher protein identification compared to a specific (non-redundant NCBI) database search alone. However, identification of a protein based on a high statistical score after MS analysis does not always provide unambiguous and accurate assignment of a specific biological function for that hit. This is because MS uses relatively few spectral information to identify the peptide, which is then matched with the computationally predicted gene and protein databases to identify the protein, while a number of peptides with a lower sensitivity are ignored from the query [5].
An important issue with BLAST and MS BLAST is the cross species protein identification; the success rate depends on the sequence identity between the query protein and its closest homologue in a database. However, the e-value of any BLAST similarity search is not always conclusive to confirm the identification of any protein or its homolog [5]. This is because it depends with the length of query sequence and therefore a specific cut-off point is difficult to determine for hundreds of searches where the query sequence string varies greatly (especially in MS BLAST approaches used in the present study where it depends on the position of peptides in a long continuous contig). During this study, the identification of a protein after BLASTp searching was based on several factors, like the number of peptides that matched with the database sequence, top hits of Cryptosporidium, the percent identity (or similarity) of the submitted query with the predicted amino acid sequences, etc. The high percent identity showed by most of the identified proteins and the 75% proteins having at least 2 identified peptides clearly indicate satisfactory level of success from MS BLAST strategy. In addition, there were a number of proteins (n = 48) for which a single matched peptide was recorded. We can assume that these are either 'true' (considering their high sequence similarity and accepting that they might be those proteins containing only few peptides) or a 'false positive' (more likely to be found in a complex mixture of peptides). Still, as the present analysis was done specifically with a C. parvum protein sample, MS-based identification of a single peptide could be used as an important aide to help identify the actual protein with very few peptides (provided the single matched entry is not a 'false positive' hit). Further complementary analyses are essential to confirm the existence of proteins for which single peptide hits were recorded.
Proteins that are evolutionarily related (i.e. have a common ancestor) are commonly referred to as homologues and very close homologues often have a similar function [26]. A homology-based functional annotation is a simple prediction method that assigns proteins that have not been annotated with the function of their annotated homologues [17]. However, it is not clear what level of sequence similarity ascertains that two proteins have the same function [18,19,23,27]. The alternative approach of structure-based function prediction also could be useful, but there are reports of unsuccessful function prediction based on sequence homology alone. In fact, although powerful for the prediction of unknown functions, homology based prediction can be notoriously inaccurate and limited in some cases [26].
Predictions of subcellular locations of identified proteins have been achieved using bioinformatic tools. The identification of the usual location of a hypothetical protein is a crucial step to identifying its role. Despite large-scale experiments involving localization in yeast, homology-based inferences are available for less than a third of all human proteins because of the lack of annotated homologues [17]. The success of various prediction methods for subcellular localisations varies. Some use signal sequences (SignalP) [2], whilst others use more generalized features, such as overall amino acid composition and predicted structural features [14]. The available tools for these predictions have some limitations, such as resolving integral membrane proteins, or proteins that have multiple locations. While some methods are successful in differentiating between membrane and non-membrane proteins [3,7,13], the prediction of all transmembrane proteins are still not reliable.
The completion of the two genome sequence projects of Cryptosporidium has contributed significantly toward their post-genomic investigation. Cryptosporidium has the most accessible Apicomplexan genome, being only 10 Mb and with relatively few introns, both of which facilitate easier gene identification. The genome sequence project of C. parvum predicted 3807 proteins from the nuclear genome [1], but the number of total proteins in the sporozoite stage is difficult to ascertain. In one study on the Plasmodium proteome (which resolved 46% of the whole P. falciparum genome), 43% of the total identified proteins were found from the sporozoite stage [4]. Considering this proportion, one can expect at least 1,700 proteins (43% of 3952 predicted proteins) in the sporozoite stage of C. parvum. However, they exclude possible PTMs which can significantly increase the actual protein species. During this study only 196 sporozoite proteins were identified and therefore remaining proteins (at least 1,500 entries) need to be resolved by further proteomic studies. With the availability of the complete genome sequences of C. parvum and C. hominis, successful characterization of their proteome is now a real possibility. The management of large computer databases are now possible and improved computational capability with efficient software has enabled us to understand the genome structure and prediction of functional proteomes [25].
Sequence similarity based searches extend the scope of proteomics in great extent. The MS BLAST search strategy has proved to be a powerful technique in identifying novel protein and peptide sequences from any organism with complete or partially sequenced genomes. In addition to other BLAST search techniques, the use of MS BLAST strategy for analyzing MS data could be useful in exploring the Cryptosporidium genome. It also can lead to the annotation of EST and genome sequences submitted in the database.



 XML Download
XML Download