

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Nicotinamide adenine dinucleotide (NAD+) functions not only as a coenzyme in oxidation-reduction reactions, but also as the adenosine diphosphate (ADP)-ribose donor in many cellular processes, such as ADP-ribosylation or sirtuin-mediated deacetylation [1]. NAD+ can be produced in human cells in two ways: the “de novo” pathway, using tryptophan as a precursor, or the “salvage” pathway, using nicotinamide (NAM), nicotinic acid, or nicotinamide riboside as precursors. The salvage pathway is the main process for NAD+ synthesis and involves the key enzyme nicotinamide phosphoribosyltransferase (NAMPT). This enzyme produces nicotinamide mononucleotide (NMN), which is subsequently converted to NAD+ by NMN adenylyltransferase in an energy-dependent reaction requiring adenosine triphosphate [2].
Owing to their high rate of proliferation and DNA synthesis, cancer cells considerably rely on NAD+-dependent signaling, and therefore, have a high rate of NAD+ turnover compared to that of normal cells. Unlike oxidative-phosphorylation systems that interconvert NAD+ and NADH, NAD+-dependent signaling pathways consume NAD+ constantly and in large amounts. This is the reason why the half-life of NAD+ is very short (about 1 hour) in cancer cells [3]. Accordingly, NAMPT is essential for the survival of tumor cells. NAMPT overexpression has been observed in several cancers, including prostate cancer, renal cancer, breast cancer, and myeloma, suggesting a key role of this enzyme in tumor biology [4]. In addition, pharmacological and genetic blockade of NAMPT reduces the viability of multiple types of cancer cells and can inhibit the growth of tumor xenografts in vivo [567]. FK866, a specific inhibitor of NAMPT, is able to reduce cellular NAD+ content, restrict cell growth, upregulate apoptotic genes (such as p53 and Bcl-2-associated X protein [BAX]), and induce apoptosis in various cancer cells [58910].
Breast cancer develops in 14% of women and is the leading cause of cancer-related death in women worldwide [11]. Understanding the molecular mechanisms of breast carcinoma progression is important for developing effective treatments. To date, NAD+-dependent signaling in breast carcinoma has been poorly investigated. One of many important ways cancer cells can rapidly divide and escape from apoptosis is by the deacetylation of proteins using NAD+. Accordingly, the aim of this study was to investigate cancer cell survival, apoptosis, and how key regulatory components are affected by NAD+ depletion in estrogen receptor (ER)-positive, p53-positive wild-type breast cancer cells. To do this, we evaluated the effects of NAMPT inhibition by FK866 in MCF-7 cells and investigated molecular pathways underlying apoptosis.
METHODS
Reagents
MCF-7, MDA-MB-231, and MCF-10 cells were obtained from the Cell Bank of the Iranian Biological Resource Center (Tehran, Iran). FK866 and NAD+ were purchased from Sigma-Aldrich (München, Germany) and dissolved and divided into aliquots, according to manufacturer instructions. Roswell Park Memorial Institute (RPMI)-1640 medium was purchased from Biowest (Paris, France), and all other cell culture reagents, including fetal bovine serum (FBS), trypsin/EDTA solution, Accutase enzyme, and antibiotics, were purchased from Gibco (New York, USA). Rabbit anti-p53, anti-acetyl-p53, and anti-β-actin antibodies were purchased from Santa Cruz Biotechnology (New York, USA). Horseradish peroxidase (HRP)-conjugated mouse or goat anti-rabbit IgG was purchased from Cell Signaling Technology (New York, USA).
Cell culture
Cells were grown in RPMI-1640 medium supplemented with 10% FBS, penicillin (100 U/mL), and streptomycin (100 µg/mL) at 37℃ in a humidified incubator with 5% CO2. Cells were detached from the culture flasks by removal of the growth medium and the addition of 1 mL trypsin/EDTA solution (0.05% w/v trypsin, 0.016% w/v EDTA). Trypsinization was stopped by the addition of culture medium containing FBS. For cell treatments, FK866 and NAD+ were used at concentrations of 10 nM and 30 µM, respectively.
MTT assay
Cells were seeded in a 96-well plate at a density of 10,000 cells/well and treated with different concentrations of FK866. After incubation for 72 hours, 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-tetrazolium bromide assay (MTT) (5 mg/mL; Sigma-Aldrich) was added to each well and cells were incubated for 3 hours. Subsequently, the medium was aspirated and dimethyl sulfoxide (DMSO) was added to dissolve the resulting formazan crystals. Next, the absorbance of the colored product was measured at 570 nm with a plate reader (BioTek Instruments Inc., Winooski, USA).
NAD+ assay
Total cellular NAD+ was measured using the colorimetric assay kit (Abcam, London, UK). Cells were seeded in 6-well plates (5×105 cells/well) in triplicate. After 24 hours of incubation, the cells were treated with FK866 for 72 hours and the total NAD+ was determined, according to the manufacturer's instructions. Briefly, cells were lysed by lysis buffer and the lysate was deproteinized by perchloric acid to avoid enzymatic digestion. Potassium hydroxide was added to neutralize the acid and balance the pH. Consequently, the lysate and standard solution were reacted with the developer in 96-well plates in triplicate. The plate was incubated at room temperature in the dark for 30 minutes following the addition of specific enzyme. Finally, the absorbance was measured at 450 nm by a plate reader and the concentration of NAD+ was determined accordingly.
Western blotting
After treatment with FK866, cells were harvested, washed with cold phosphate-buffered saline, and lysed in buffer containing 1% Triton X-100, 20 mM Tris-HCl, and phosphatase and protease inhibitors (sodium orthovanadate, sodium fluoride, aprotinin, leupeptin, and phenylmethane sulfonyl fluoride, pH=8.3). Concentrations of the protein samples were determined with a micro BCA protein assay kit (Thermo Fisher Scientific, Waltham, USA). Samples were then loaded and resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and proteins were transferred to polyvinylidene difluoride membranes. Membrane blocking was performed using 5% bovine serum albumin overnight. Primary antibodies against p53, acetylated p53, NAMPT, and β-actin were used at a 1:1,000 dilution and HRP-conjugated secondary antibodies were used at a 1:10,000 dilution. Immunoreactive bands were visualized using chemiluminescence HRP substrate (GE Amersham, London, UK). Quantification was performed using ImageJ (NIH, Bethesda, USA) and results were normalized to the β-actin band intensity as internal control for loading variations.
Real-time polymerase chain reaction
Total RNA was extracted from MCF-7 cells using a Hybrid-R RNA purification kit (GeneAll Biotechnology, Seoul, Korea). The quality and quantity of RNA were analyzed using a NanoDrop spectrophotometer (NanoDrop Technologies, Wilmington, USA). Consequently, cDNA (0.5 µg) was synthesized using a RevertAid RT kit (Thermo Fisher Scientific). Real-time polymerase chain reaction (PCR) was carried out using a SYBR green kit (TaKaRa, Tokyo, Japan) with gene-specific primers. Primer sequences for p21, BAX, and β-actin were as follows: p21: 5′-GCTCGGCTCTTCACCAAG-3′ (forward), 5′-GTCACTGTCTTGTACCCTTGTG-3′ (reverse); BAX: 5′-GGGTGGTTGGGTGAGACTC-3′ (forward), 5′-AGACACGTAAGGAAAACGCATTA-3′ (reverse); ACTB: 5′-GGTGGCTTTTAGGATGGCAAG-3′ (forward), 5′-ACTGGAACGGTGAAGGTGACAG-3′ (reverse). Primers were designed by Oligo 7 software (Molecular Biology Insights Inc., Colorado Springs, USA) and their specificities were checked using primer-BLAST (NCBI, Bethesda, USA; http://blast.ncbi.nlm.nih.gov/Blast.cgi). β-Actin was used as a normalization control. The standard PCR conditions were: 95℃ for 15 minutes, followed by 40 cycles at 95℃ for 5 seconds, 60℃ for 30 seconds, and 72℃ for 40 seconds. Specificity was verified by a dissociation curve for each set of primers with increasing temperature from 60℃ to 95℃. Standard curves for each primer were generated from serial dilutions of a pooled sample of cDNA. Statistical analyses of the PCR results were performed using the Rotor-Gene System Software (Qiagen, Hilden, Germany) based on the 2-ΔΔCT method, which calculated relative changes in gene expression of the target gene normalized to β-actin. These experiments were repeated at least three times independently.
Apoptosis assay
Apoptotic cells were assessed by an Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI) kit (Roche Applied Science, Mannheim, Germany). Cells were seeded at a density of 3×105 cells per 6-well plate and treated with 10 nM of FK866 for 36 hours. Treated cells were harvested using accutase enzyme, washed twice with ice-cold phosphate-buffered saline, resuspended in a master mix containing reaction buffer, Annexin V-FITC, and PI, and incubated for 15 minutes at room temperature (25℃) in the dark. Samples were immediately analyzed by a flow cytometer (BD Biosciences, San Jose, USA) using a laser at 488 nm excitation with a bandpass filter at 515 nm and 600 nm for FITC and PI detection, respectively.
Statistical analysis
Data from three or more independent experiments were presented as means±standard error of the mean. Statistical analyses were performed by one-way ANOVA, followed by Tukey's post-hoc test using SPSS software version 16 (SPSS Inc., Chicago, USA). For all analyses, p-values less than 0.05 were considered statistically significant.
RESULTS
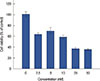
FK866 reduced MCF-7 cell survival
Using an MTT cell viability assay, the cytotoxic effects of FK866 on MCF-7 cells were observed. FK866 significantly reduced cell viability in a concentration-dependent manner (Figure 1). A 10-µM concentration inhibited 41% of cell growth.
FK866 reduced intracellular NAD+ levels
NAMPT was highly expressed in MCF-7 cells compared to the levels in MCF-10 or MDA-MB-231 cells (Figure 2A). MCF-7 cells were treated either with FK866 (10 nM) or DMSO (control). All samples used in the assay had equal protein concentrations. As shown in Figure 2B, cells exposed to 10 nM of FK866 underwent ~50% NAD+ depletion (p<0.001), compared to control cells.
FK866 increased the levels of p53 and its acetylated form
Since acetylation of p53 is an NAD+-dependent process and to further address the mechanism of cell cycle arrest and apoptosis induced by NAD+ depletion, we examined the effect of FK866 on the acetylation status of p53 in MCF-7 cell line by Western blotting (Figure 3). Significant increases in the total and acetylated p53 levels were observed in FK866-treated cells, compared to the levels in control cells (Figure 3A and 3B, respectively). Addition of exogenous NAD+ completely abolished the effect of FK866 on p53 and its acetylation levels (Figure 3A and 3B).
FK866 induced p21 and BAX gene expression
The mRNA expression of p21, an important cell cycle inhibitor, and BAX, a protein involved in apoptosis, were measured in MCF-7 and MDA-MB-231 cells. The mRNA expression levels of both of these genes were increased 72 hours after treatment with FK866 in MCF-7 cells (Figure 4A and 4B) and in MDA-MB-231 cells (Figure 4C and 4D). Again, the addition of exogenous NAD+ prevented the upregulation of p21 and BAX.
FK866 induced cell apoptosis
A flow cytometry apoptosis assay was performed to check the efficiency of FK866 in MCF-7 cells to induce apoptosis and confirm previous findings. The flow cytometric data shown in Figure 5A and its quantification in Figure 5B showed FK866 significantly increased the level of programmed cell death in MCF-7 cells. Addition of exogenous NAD+ reversed the effects of FK866 on apoptosis, indicating that FK866 could induce apoptosis via NAD+ depletion.
DISCUSSION
Cancer cells have an increased turnover of NAD+, a coenzyme involved in multiple metabolic reactions and cell signaling pathways [12]. In mammals, NAD+ is mainly produced via the salvage pathway, which uses NAMPT as a key regulatory enzyme [2]. Recently, NAMPT has attracted attention as a potential therapeutic target in cancers and other metabolic diseases [13]. It has been shown that physiological functions of NAMPT are conserved in different types of cancer cells, and thus, NAMPT inhibitors may have clinical value for the treatment of a variety of different types of cancers [14]. In many studies, pharmacological or genetic blockade of NAMPT leads to reduced cellular NAD+ levels, upregulated levels of apoptotic genes, and apoptosis of cancer cells [56715]. FK866, which is a highly specific noncompetitive inhibitor of NAMPT [5] has been shown to have beneficial effects in triple-negative breast cancer cells [16]. In the management of ER- and human epidermal growth factor receptor 2-positive breast cancers, endocrine therapy is most likely considered as the first-line of therapy [17]. However, ER-positive breast cancers with poor prognoses require more aggressive therapies and are recommended to be treated with chemotherapy, even in the case of ER-positive diseases [18]. Zhang et al. [19] suggested that MCF-7 cells, which are an ER-positive cell line, benefit from NAD+ depletion by genetic inhibition of NAMPT. However, the effect of NAMPT inhibition by FK866 has not been previously studied in ER-positive breast cancer cells.
Our results showed that FK866 was able to reduce the cellular NAD+ content in MCF-7 cells, and subsequently, increase the activity of p53, induce p21 and BAX expression, and eventually, induce apoptosis. FK866 increased total p53 and acetylated-p53 levels, while the addition of exogenous NAD+ abolished this effect. It is currently known that eight lysine residues on the p53 protein can be acetylated [20]. Previous studies suggest that in the presence of different extracellular stresses, acetylation of p53 helps to better coordinate p53-mediated downstream signaling [21]. Sirtuin-1 can effectively deacetylate p53 and attenuate p53-mediated functions using NAD+ via NAMPT production [22]. Therefore, NAD+ depletion by FK866 could have prevented p53 deacetylation and stabilized its acetylated form. Our findings were supported by previous studies that demonstrated the strong relationship between NAMPT activity and p53 acetylation and downstream effects [8]. It is worthy to note that p53 acetylation itself is effective in stabilizing and increasing the levels of total p53 [23]. Thus, it is plausible that with the treatment of FK866, p53 protein levels increased concomitant with p53 acetylation.
The tumor suppressor protein p53 affects a vast number of downstream targets, which are involved in cell cycle arrest and apoptosis. Among them are p21 and BAX, which are major mediators of p53 function [24]. Previous studies performed on other cell lines have shown that NAMPT inhibition results in the upregulation of p21 and BAX expression via NAD+ depletion [58102526]. In our study, FK866 increased p21 and BAX expression, confirming p53 activation. Thus, FK866 treatment could successfully induce molecular mediators of cell cycle arrest and apoptosis. Conversely, increased cellular NAD+ levels prevented FK866-induced upregulation of p21 and BAX, further emphasizing the involvement of NAD+ in cell cycle progression and apoptosis. The activation of p21 and BAX occurrs via both p53-dependant and p53-independent mechanisms. For example, FOXO1, which is a target of sirtuins for deacetylation, can upregulate p21 and BAX [272829]. Thus, increased cellular NAD+ levels could lead to p21 and BAX upregulation independent of the p53 status, as it was observed in MDA-MB-231 cells that have a mutated p53.
Apoptosis plays an important role in cancer treatment as it is a popular target of many treatment strategies [30]. We measured early apoptosis in cells treated with FK866 compared to that of control cells treated with DMSO. FK866 induced a significant increase in apoptosis compared to control conditions, while addition of exogenous NAD+ prevented FK866-induced apoptosis.
The results of the present study demonstrate that NAMPT had a key role in cell survival and evasion of apoptosis in MCF-7 breast cancer cells by means of its product NAD+. Furthermore, chemical inhibition of NAMPT was an effective tool to reduce NAD+ production, upregulate p53 activity, and induce apoptosis in these breast cancer cells.



 XML Download
XML Download