

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Male breast cancer (MBC) is an extremely rare disease, accounting for less than 1% of all diagnosed breast cancer cases and cancer cases in men [12]. MBC accounts for only 0.7% of all male cancers in India [3]. Although it has traditionally been considered to be very similar to postmenopausal female breast cancer, recent research suggests that MBC has distinct histological and molecular profiles [4]. Interestingly, a recently published report covering more than 40 years of data indicated that males with breast cancer were at increased risk of death and disease recurrence compared to their female counterparts [5]. Therefore, there is a need for more thorough studies of MBC, particularly for underrepresented populations such as those in developing nations.
CASE REPORT
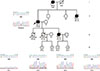
An Indian male farmer aged 72 years was referred to Mehdi Nawaz Jung Institute of Oncology and Regional Research Centre, Hyderabad, India in May 2010 with a complaint of a lump in the right breast. The patient revealed a strong family history of breast cancer; his mother, sister, and only daughter all died from the disease (Figure 1). He was an ex-smoker with 20 pack years. He was married to a woman with no history of breast or any other cancer and had two sons and a daughter. Informed consent was obtained from the patient and family members before participation in the study. The study was approved by the Institutional Ethics Committee (No. 891A/IG/IEC/2011).
The patient had no other comorbidities, and his physical, biochemical, and hematological blood tests were all within the normal range. Fine needle aspiration cytology was initially performed to confirm the diagnosis, followed by a modified radical mastectomy, which was subjected to histopathological examination. The tumor size was 3.5×3×3 cm, with significant invasion of the nipple, and 11 of 12 lymph nodes tested positive for malignancy. The diagnosis was infiltrating ductal carcinoma T4bN3M0. He received six cycles of adjuvant chemotherapy (1,000 mg fluorouracil, 100 mg adriamycin, and 100 mg cyclophosphamide) at 21-day intervals and adjuvant radiotherapy (50 Gy), followed by tamoxifen (20 mg once daily) given that the tumor was positive for estrogen receptor/progesterone receptor (>90%) and negative for human epidermal growth factor receptor expression. The BRCA1 and BRCA2 sequences were normal with no mutations detected. He was found to be disease-free after 30 months of follow-up.
Because of the rarity of MBC and the strong family history of breast cancer in this case, blood samples were collected from the patient and his four grandchildren (the only surviving dependents). Roughly one in three MBC diagnoses occur in families with an incidence of breast and ovarian cancer [6]. Genetic tests determined that most of the genes typically associated with MBC (BRCA1/2, TP53, CHK2, PALB2, and ATM) were normal in this patient. After learning of the patient's family history of breast cancer, we further tested the moderate breast cancer penetrant gene BRCA1-interacting protein 1 (BRIP1, also known as BACH1 and FANCJ) located on chromosome 21 [7]. Polymerase chain reaction primers were designed to amplify all 19 exons that encode the BRIP1 gene from genomic DNA isolated from the blood of the patient and all four surviving descendants. Nested primers were designed and each exon was sequenced from both directions using the ABI Prism Big Dye Terminator cycle sequencing kit (Applied Biosystems, Foster City, USA) on the ABI 3730 sequencer (Life Technologies, Carlsbad, USA). Two BRIP1 mutations were identified in the patient (Figure 2). The first is a homozygous mutation from guanine to adenine at nucleotide 2637; however, this is a silent mutation that does not result in an amino acid change in the protein (E879E). The resulting nucleotide triplet (GAA) is used less frequently than the wild-type, but is still relatively common and would not be expected to alter translational efficiency. The second mutation resulted in a change from cytosine to thymine at position 2755 that converts the normal proline at residue 919 to a serine. The patient was found to be heterozygous for this P919S missense mutation, with one allele altered and the other remaining the wild-type. Analysis of the patient's four surviving descendants (all grandchildren) revealed that all four were also heterozygous for the P919S mutation. Interestingly, two were also heterozygous (Figure 1, individuals 3 and 4) for the E879E silent mutation and the other two had the wild-type sequence at this location (Figure 1, individuals 2 and 5). However, the discrepancy in the inheritance of these mutations cannot be explained due to the lack of genetic information from the other family members. It is interesting to note that both of these mutations are located in exon 19 and are quite close together in both the nucleotide sequence and the protein.
The P919S mutation was analyzed using the SIFT (http://sift.jcvi.org) predictive algorithm for amino acid substitution and functional prediction [8]. A score of less than 0.05 indicates that a particular amino acid substitution is predicted to alter the protein structure. The score for the P919S mutation was 0.28 and is therefore expected to be tolerated. However, this residue is located in the vicinity of the BRCA1-binding domain, and therefore mutations in this region could be particularly damaging. Figure 2 shows other BRIP mutations located near position 919 that have been previously associated with breast cancer.
DISCUSSION
Although the frequency of female breast cancer has been decreasing since the late 1990s, analysis of SEER data shows an increase in the incidence of MBC with peaks in 2011 and 2012, the last 2 years for which data are available [9]. Therefore, it is critically important to analyze mutations associated with MBC separately from those linked to female breast cancer. This is particularly important in ethnic groups/nationalities that have typically been neglected in these types of studies. Here, we have presented the case of an MBC patient with a strong familial history of breast cancer that was wild-type for BRCA1/2 and other genes commonly associated with breast cancer. However, the patient was found to have two mutations in BRIP1. The tumor suppressor BRIP1 (synonyms: BACH1 or FANCJ) is a DNA helicase that is commonly mutated in Fanconi anemia and certain cancers. BRIP1 interacts with BRCA1 protein, a known tumor suppressor, and truncations in the BRIP1 gene doubles the risk for developing breast cancer [10]. Although BRIP1 and several of its mutations have been studied in female breast cancer, major differences between male and female breast cancers have recently come to light [4]; therefore, the role of BRIP1 in MBC requires further evaluation. Consistent with its role as a tumor suppressor, BRIP1 is a regulator of the DNA damage response and is important for maintaining chromosomal stability [1112]. However, the specific mutations associated with these conditions have not been elucidated.
Previous studies have examined the relationship between the heterozygous P919S mutation and breast cancer; however, these studies were only conducted in European populations and the findings were contradictory. The first study showed a clear association between P919S and an increased (4.5-fold) risk of early-onset breast cancer (before the age of 50) [13], whereas the other two studies did not detect a correlation between P919S and breast cancer [1415]. This lack of agreement between studies indicates a need for more studies related to this mutation, and could be due to ethnic variations between populations.
Analysis of the P919S mutation with SIFT [10] showed that it is unlikely to alter protein structure; however, BRIP1 interacts with BRCA1 [7] through the BRCT domains located in the vicinity of this mutated residue. Nevertheless, without a comprehensive molecular characterization it is difficult to conclude whether this mutation affects these protein interactions. It is important to note that all four of the grandchildren in the family carry the heterozygous P919S mutation and none is homozygous for the E879E silent mutation. However, most of these grandchildren are in their early 20s; therefore, large-scale genetic screens in this ethnic population are necessary to predict whether these variants increase the breast cancer susceptibility. Such studies could potentially provide new insights into the proper management of the disease and plan for appropriate preventive and therapeutic intervention.
In summary, this report presents a rare case of MBC with a significant history of familial breast cancer, resulting in the deaths of many of the women in the family. Therefore, reports of this kind are important for understanding the influence of genetic variants in different ethnic groups. Moreover, to the best of our knowledge, this is the first study to detect mutations in BRIP1 linked to MBC in the Indian population.

 XML Download
XML Download