

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Silibinin (silybin), a polyphenolic flavonoid [1], is the major active component of silymarin, and is extracted from milk thistle (Silybum marianum). Silibinin has hepatoprotective, anti-inflammatory, and antioxidant properties [2]. Consuming silibinin is safe and nontoxic to animals and humans. Also, the anticancer activities of silibinin have been revealed in various epithelial cancers such as prostate, colon, skin [2], bladder, and ovarian cancer [1]. Due to its chemopreventive and chemotherapeutic properties, silibinin is currently in phase I/II clinical trials for treatment of prostate cancer. It has been demonstrated that silibinin decreases the expression of antiapoptotic Bcl-2, and increases the expression of proapoptotic Bcl2-associated X (Bax) and caspase 8 (Casp-8), in MCF-7 and T47D breast cancer cells [1]. Thus, silibinin likely induces apoptosis in breast cancer through both the intrinsic and extrinsic pathways. However, further studies are needed to fully understand the mechanism by which this occurs.
MicroRNAs (miRNAs) are 22 nucleotide long RNAs that negatively regulate gene expression by binding to their complementary target mRNAs and inducing their degradation or repressing their translation [3]. Bioinformatic analyses have predicted that miRNAs regulate the translation of more than one-third of all human protein-encoding genes [4]. In silico approaches (online programs such as TargetScan and miRWalk) can be applied to predict potential miRNA targets and their related signaling pathways [5]. miRNAs are implicated in cellular processes such as apoptosis, cell differentiation, cell proliferation and tumor suppression [36]. Recent studies have shown that miRNAs play a critical role in cancer development and progression [6]. The aberrant expression of miRNAs or their mutation has been associated with different stages of cancer [78]. Indeed, miRNAs can act as tumor suppressors or oncogenes. miR-21 and miR-155 are two oncomiRs [6] that are commonly upregulated in a number of cancers such as breast, lung and colon cancers [7]. Hence, these miRNAs are potential candidates for cancer diagnosis and therapy. The upregulation of miR-21 and miR-155 in a variety of cancer cells prompted us to investigate the correlation between silibinin treatment and the expression of these oncomiRs in MCF-7 cells. Our results showed that silibinin induces cell death by downregulating miR-21 and miR-155. Furthermore, a quantitative analysis demonstrated that silibinin induces apoptosis in MCF-7 cells through the regulation of genes from both the extrinsic and intrinsic pathways.
METHODS
Cell culture
The MCF-7 (adenocarcinoma) human breast cancer cell line was purchased from the National Cell Bank of Iran (NCBI, Pasteur Institute of Iran). The cells were cultured in RPMI1640 media supplemented 10% fetal bovine serum antibiotics (100 U/mL penicillin and 100 µg/mL streptomycin) and glutamine (2 mmol/L), at 37℃ in a humidified atmosphere containing 5% CO2.
Cell proliferation assay
To determine the effect of silibinin on cell proliferation, an 3-(4,5-dimethylthiazol-2-yl) 2,5-diphenyl tetrazolium bromide (MTT) assay was performed. Briefly, 7×103 cells/well were seeded in 96-well plates and treated with different concentrations of silibinin (0–300 µM; Sigma Aldrich, Deisenhofen, Germany) for 24, 48, or 72 hours. Then, MTT dye (0.5 mg/mL; Sigma Aldrich) was added to the wells and incubated at 37℃. The formazan crystals were dissolved by adding dimethyl sulfoxide (DMSO; 100 µL/well), and the optical density was measured at 570 nm using an enzyme-linked immunosorbent assay microplate reader. Each experiment was performed a minimum of three times.
Cell cycle assay
Cell cycle analysis was performed by flow cytometry. Treated cells were harvested, then washed with phosphate buffered saline, fixed in 70% ethanol and stored at -20℃ for over 2 hours. The fixed cells were resuspended in propidium iodide (PI; Sigma Aldrich) containing 0.1% (v/v) Triton X-100 and 2 mg DNase-free RNase A (Thermo Fisher Scientific Biosciences GmbH, St. Leon-Rot, Germany). Stained cells were incubated for 15 minutes at 37℃ prior to flow cytometric analysis using the CyFlow®-SL system (Partec GmbH, Münster, Germany).
Quantitative real-time polymerase chain reaction analysis of miRNA expression
RNA extraction was performed using the miRCURY™ RNA isolation kit (Exiqon, Vedbaek, Denmark) according to the manufacturer's instructions. The concentration of RNA was determined using a NanoDrop 1000 (Thermo Scientific, Wilmington, USA). Complementary DNA (cDNA) was synthesized using the miR-Amp kit (Parsgenome, Tehran, Iran). First, a poly-A tail was added to the extracted RNA by poly(A) polymerase at 37℃. The RNA was then mixed with reverse transcriptase, reaction buffer, and miRNA specific primers. These primers are comprised of oligo-dT and some specific nucleotides complemented with considered miRNA that in quantitative real-time polymerase chain reaction (qRT-PCR) targeted by forward or reverse primer as template. This mix was incubated for 60 minutes at 45℃ and inactivated for 1 minute at 85℃ to obtain the cDNA. qRT-PCR was performed by SYBR® Premix Ex Taq™ II (Takara Bio, Shiga, Japan) and performed with an Applied Biosystems StepOne™ instrument (Applied Biosystems, Foster City, USA) programed as follows: 95℃ for 10 seconds, followed by 40 cycles at 95℃ for 5 seconds, 62℃ for 20 seconds, and 72℃ for 30 seconds. The amount of each miRNA was normalized relative to the amount of U6 small nuclear RNA. The primer pairs were obtained from Parsgenome. The qRT-PCR analyses were performed in triplicate. The fold-change of each miRNA was calculated using the 2-ΔΔCt method.
Prediction of miRNA target genes
A bioinformatics approach was used to identify potential miR-21 and miR-155 targets. Targets within the apoptotic pathways were predicted using several online algorithms such as TargetScan (http://www.targetscan.org/), miRWalk (http://www.umm.uni-heidelberg.de/apps/zmf/mirwalk/), and Diana microT (http://diana.imis.athena-innovation.gr/). These algorithms evaluate potential miRNA targets based on the presence of complementary binding sites in their 3' untranslated region (3'-UTR) and the minimum binding energies, to determine whether the interaction between the miRNAs and the targets are thermodynamically favorable [9]. To avoid overprediction, the results coming from the different databases were intersected and only those genes predicted by at least four of the algorithms were considered.
Quantitative analysis of target genes
The transcription levels of the potential miR-21 and miR-155 target genes, apoptotic peptidase activating factor 1 (APAF-1), caspase 9 (CASP-9), BH3 interacting domain death agonist (BID) and p53, were evaluated by qRT-PCR. Briefly, miRCURY™ RNA Isolation kit (Exiqon) was used for RNA extraction. CDNA was synthesized using the PrimeScript™ RT reagent Kit (Takara Bio) according to the manufacturer's instructions. Quantitative analysis of target gene expression was performed with SYBR® Premix Ex Taq™ II (Takara Bio), using the Applied Biosystems StepOne™ instrument. PCRs were performed by heating to 95℃ for 15 seconds, followed by 40 cycles at 95℃ for 5 seconds and 60℃ for 30 seconds. The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was used as an endogenous internal control. The primers used are listed in Table 1. All reactions were run in triplicate and the fold-change of each gene was calculated by the 2-ΔΔCt method.
Statistical analysis
To test for statistical significance, Kruskal-Wallis testing with Dunn's multiple comparison test for post hoc analysis was performed. The results are presented as mean±standard deviation. Student t-test and one-way ANOVA were used to analyze significance differences between the two cell groups (treated and untreated). A p-value of 0.05 was used to determine statistical significance. Each experiment was performed in triplicate. All statistical calculations were performed using the SPSS software, version 16.0 (SPSS Inc., Chicago, USA).
RESULTS
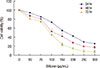
Decreased proliferation in silibinin-treated MCF-7 cells
MCF-7 cells were treated with different concentration of silibinin for 24, 48, or 72 hours. The MTT assays (Figure 1) revealed that silibinin decreased proliferation and cell viability in a dose- and time-dependent manner. Our analyses determined that silibinin (100 µg/mL) decreased cell viability to ~50% after 48 hours. However, in the same amount of time, treatment with 150 µg/mL of silibinin decreased viability to ~40% (p<0.001) compared to untreated control.
Increase in the sub-G1 population of silibinin-treated MCF-7 cells
Decreased cell viability was observed by MTT assay after 48 hours in cells treated with silibinin (100 or 150 µg/mL) compared to untreated cells. To identify the correct silibinin concentration (100 or 150 µg/mL) for gene expression analysis, the percentage of cells in the sub-G1 phase of the cell cycle was measured by PI staining after silibinin treatment for 24 or 48 hours (Figure 2). Flow cytometric analysis showed that silibinin-treated cells suffered a greater degree of death on day 2 than day 1. Furthermore, cell viability decreased below 40% with 48 hours high dose silibinin treatment (150 µg/mL), and the sub-G1 population was too extensive for these cells to be used in gene expression analysis. Therefore, we selected 48 hours lower dose silibinin (100 µg/mL) treatment for further experiments.
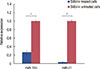
Downregulation of miR-21 and miR-155 after silibinin induction
To further investigate the anticancer activity of silibinin, the expression levels of two oncomiRs (miR-21 and miR-155) were evaluated by qRT-PCR in silibinin-treated (100 µg/mL) and untreated MCF-7 cells. Quantitative analysis demonstrated that the expression levels of miR-21 and miR-155 had significantly decreased (p<0.05) after 48 hours silibinin treatment (Figure 3).
Potential targets of miR-21 and miR-155 within apoptotic pathways
To investigate the function of miR-21 and miR-155 in silibinin-treated cells, in silico predictions were used to determine their potential targets within the intrinsic and extrinsic apoptosis pathways. Among the potential targets listed in Table 2, the most interesting and promising targets include CASP-9, BID, APAF-1, CASP-3, CASP-6, CASP-8, CASP-10, FAS-L, BCL2L15, BCL2L2, and programmed cell death 4 (PDCD4), as they were each predicted by a minimum of four algorithms.
Silibinin promotes apoptosis through regulation of downstream apoptotic genes
To further investigate the in silico predictions, the expression levels of four potential targets were quantitatively analyzed in silibinin-treated and untreated cells. As shown in Figure 4, after 48 hours, CASP-9, p53, and BID levels were upregulated by 4.2±0.41, 6±0.43, and 1.6±0.26 fold, respectively, in silibinin-treated cells compared to untreated cells. The expression of APAF-1 decreased with silibinin treatment.
DISCUSSION
The antitumor property of silibinin has been demonstrated in various types of cancers such as lung, bladder, hepatoma, and prostate carcinomas. Also, several studies have reported upregulation of the numerous genes related to apoptosis or cell cycle arrest after silibinin treatment [110]. However, the role of miRNAs in silibinin treatment remains unclear [11]. Nowadays, it is recognized that miRNAs are implicated in multiple biological pathways such as cell cycle regulation, tumor suppression, apoptosis and cell proliferation [5]. Dysregulation of miRNAs and their target genes is associated with uncontrolled cell growth and tumorigenesis [12]. In our study, we assessed the expression of miR-21 and miR-155, two known oncomiRs, as well as that of their potential targets, in silibinin-treated T47D [13] and MCF-7 cells. Our results show that silibinin diminishes proliferation and increased apoptosis in a dose- and time-dependent manner in both silibinin-treated T47D [13] and MCF-7 cells.
As expected, MCF-7 cells treated with silibinin (100 µg/mL) displayed downregulated miR-21 and miR-155 levels that were associated with increased apoptosis and decreased cell number. The overexpression of miR-21 and miR-155 has been previously demonstrated in some human cancers, including breast cancer [7]. Furthermore, other studies have shown that the dysregulation of miR-21 [712] and miR-155 [7] correlates with tumor progression. A study has shown a greater than twofold upregulation of miR-21 and miR-155 in breast cancer samples compared to normal adjacent tumor tissue [7]. In another study, the expression of miR-21 was evaluated in 89 breast cancer patients and 55 healthy controls. The expression level of miR-21 in breast cancer tissue was 3.39-fold higher than in the healthy controls [14]. Overexpression of miR-21 has been found in pregnancy-associated breast cancer tumors, with average levels being 11.1-fold compared to 1.8-fold in the matched normal adjacent tissue [15]. In MCF-7 and MDAMB-231 breast cancer cells, miR-21 knockdown cause the inhibition of proliferation and induction of apoptosis [616]. In addition, it has been reported that miR-155 expression is significantly greater in estrogen receptor 1 positive (ERα+) breast cancer cells (such as MCF-7 cells) than ERα negative (ERα–) cells [8]. In another study, the expression of miR-155 in MCF-7 cells was 3.05-fold higher than that in MDA-MB-231 cells [17]. In addition, miR-155 upregulation in MCF-7 cells inhibited apoptosis and stimulated cell growth [17]. Another study has shown that the overexpression of miR-155 increased the proliferation of MDA-MB-231 and MCF-7 cells, while, anti-miR-155 significantly reduced MDA-MB-231 cell growth [18]. Furthermore, anti-miR-21 inhibited the proliferation of MCF-7 and MDA-MB-231 cells in vitro, as well as tumor growth in nude mice [16]. Therefore, it is predicted that in various cancerous cells antitumor drugs, such as silibinin, could induce apoptosis through suppression of these oncomiRs. In fact, it was previously shown that treatment of erlotinib-refractory tumors with silibinin decreases miR-21 expression [11].
Recently, it has been recognized that miR-21 represses tumor suppressors and apoptotic genes including PDCD4, tropomyosin 1 (TPM1), phosphatase and tensin homolog (PTEN) [19], and Fas ligand (FASL) [20]. Downregulation or loss of PDCD4 has been observed in several tumors such as lung, renal, skin and breast tumors [21]. PDCD4 is a suppressor of cell growth that is upregulated during apoptosis, and it was demonstrated that its expression is increased upon miR-21 inhibition in MCF-7 cells [19]. TPM1 is a tumor suppressor implicated in cell migration. It appears that miR-21 plays a role in cell invasion and tumor metastasis by targeting tumor/metastasis suppressors [12]. Through the repression of PTEN, miR-21 caused v-akt murine thymoma viral oncogene homolog (AKT) activation and subsequent cell proliferation. Furthermore, in MCF-7 cells, it has been reported that miR-21 inhibits FASL expression and cell death [20]. Additionally, miR-155 targets genes such as suppressor of cytokine signaling 1 (SOCS1) and RAD51 recombinase (RAD51) [22] in breast cancer cells. SOCS1 is a negative regulator of the janus kinase (JAK)/STAT signaling pathway, subsequently activating signal transducer and activator of transcription 3 (STAT3). Activated STAT3 regulates cell survival, proliferation, and metastatic potential in breast cancer cells [18]. Thus miR-155 through SOCS1 repression increases cell proliferation. MiR-155 also represses RAD51 expression, which plays a role in DNA repair, and its dysregulation can be associated with aberrant genome rearrangements [22]. Nevertheless, the function of most of miRNAs within different cell and tissue types requires further investigation.
According to computational predictions, it seems that miRNAs regulate 30% of the protein-coding genes in the human genome [23]. Herein, to investigate the antitumor effects of miR-21 and miR-155, several algorithms including TargetScan, Diana microT, and miRWalk were used. Potential miRNA target apoptotic genes such as CASP-9, BID, PDCD4, and APAF-1 were found using the algorithms. We speculated that the downregulation of miR-21 and miR-155 in silibinin-treated cells was associated with an upregulation of their predicted targets. Our quantitative analyses showed that CASP-9 was upregulated more than 4-fold in silibinin-treated MCF-7 cells relative to untreated cells. CASP-9 is a critical component of the intrinsic apoptosis pathway and is released from mitochondria following cytochrome c release. Together with APAF-1 and cytochrome c, CASP-9 is recruited to form the apoptosome. CASP-9 then activates CASP-3 and CASP-7 leading to apoptosis [24]. Upregulation of CASP-9 suggests greater induction of the intrinsic apoptosis pathway in silibinin-treated cells. While in untreated MCF-7 cells, miR-21 and miR-155 would play a role in inhibiting the intrinsic apoptosis pathway through CASP-9 targeting.
Given that there was -10% greater cell death in silibinin-treated cells than untreated cells after 48 hours, it can be said that silibinin induces cell death. The apoptosis that occurred in untreated cells was the result of space- and other parameter-limitations. Thus, it is reasonable that treated and untreated cells underwent cell death at different rates. The downregulation of APAF-1 in treated cells compared to untreated cells may be due to the difference in apoptotic stages between the two cell groups at the time of the analysis. Nonetheless, APAF-1 has an essential role in the intrinsic apoptosis pathway and it would have been expected that APAF-1 be upregulated in silibinin-treated cells. A previous study has shown that miR-21 inhibition led to APAF-1 overexpression, confirming our targets prediction. However, in the present study, APAF-1 transcriptional levels were detected later than the other genes by the ABI StepOne approach. Therefore, its expression levels in both treated and untreated cells suffered death was fewer than other genes. It is possible that APAF-1 expression increased in treated cells later (after 48 hours) or sooner (before 48 hours) than in untreated cells, causing this discrepancy. APAF-1 function is dependent on positive and negative regulators. For example, acidic nuclear phosphoprotein 32 family members (ANP32A or PHAPI), breast cancer antiestrogen resistance 1 (BCAR1 or CAS), and heat shock protein 70-4 (Hsp70) promote nucleotide exchange on APAF-1 and prevent APAF-1/cytochrome c inactivation [25]. It is likely that in treated cells, posttranslational regulation of APAF-1 protein by enhancers plays an important role in apoptosis induction, and this possibility would need to be addressed in future work. Overall, we think that miR-21 and miR-155 are able to inhibit APAF1, but further studies are needed to confirm this hypothesis.
Recently, inhibition of CASP-8 (a component of extrinsic pathway) and CASP-9 abrogated caspase 3 (CASP-3) activity has been reported in silibinin-treated MCF-7 and T47D cells. Therefore, in breast cancer cells, silibinin induces apoptosis through mitochondrial (intrinsic) and the death receptor (extrinsic) pathways [1]. Although we did not investigate genes involved in the extrinsic pathway, overexpression of BID implies that silibinin induced apoptosis through both the intrinsic and extrinsic pathways. There is crosstalk between both pathways mediated by B-cell CLL/lymphoma 2 (BCL-2) family member BID. BID is a proapoptotic member of BCL-2 family that is cleaved by activated CASP-8 (from extrinsic pathway) [26], then truncated BID (tBID) translocates to the mitochondria and induces cytochrome c release [27]. BID expression was significantly increased (1.6-fold) in silibinin-treated cells compared to untreated cells. Therefore, it seems that both the extrinsic and intrinsic pathways play a role in apoptosis in MCF-7 cells. Considering that BID is a potential target of miR-21 and miR-155, MCF-7 cells may have a suppressed intrinsic pathway through BID inhibition.
The p53 gene was a predicted target of miR-21 and was upregulated after silibinin treatment. The p53 protein can induce the expression of several pro-apoptotic genes, which can initiate the extrinsic and intrinsic apoptotic pathways [28]. p53-deficient cells are less susceptible to apoptosis induced by silibinin [2]. Downregulation of miR-21 leads to the activation of the p53 pathway through PTEN elevation. Also, miR-21 potentially targets several transcriptional activators involved in this pathway, including junction mediating and regulatory protein (JMY), topoisomerase I binding RING Finger Protein (TOPORS), tumor protein p53 binding protein 2 (TP53BP2) and death-domain associated protein (DAXX) in transcriptional level. In glioblastoma cells, the expression of these activators was increased after miR-21 downregulation. In breast cancer cells, inhibition of miR-21 induced the expression of several genes regulated by p53 such as APAF-1, Fas cell surface death receptor (FAS) and cyclin-dependent kinase inhibitor 1A (CDKN1A or P21) [23]. Therefore, it seems that silibinin, through the downregulation of miR-21, induces the p53 pathway, which subsequently activates several genes related to the extrinsic and intrinsic apoptotic pathways.
A study has reported that in glioblastoma cells miR-21 inhibition by antisense oligonucleotides led to the activation of caspases and increased cell death [29]. Also, miR-155 knockdowns in liposarcomas induced G1-S arrest and inhibited tumor growth [30]. Taken together, it is suggested that silibinin may induce apoptosis and cell cycle arrest, in part by inhibiting the miR-21 and miR-155 oncomiRs.
In this study, the expression patterns of miR-21 and miR-155 in MCF-7 cells were evaluated after silibinin treatment. The decline in miR-21 and miR-155 levels, and subsequent increase in the expression of their potential targets (CASP-9 and BID), was correlated with the induction of apoptosis.




 XML Download
XML Download