

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Proto-oncogene B-cell lymphoma 6 (BCL6) is the master regulator of B-lymphocyte development. BCL6 promotes the proliferation of B-lymphocytes and blocks their differentiation into plasma and memory cells [1]. A study showed that high BCL6 expression is associated with tumor size, lymph node metastasis, advanced clinical stage, high tumor grade, Ki-67 labeling index, and poor prognosis in patients with breast cancer [2]. Moreover, BCL6 stimulates the oncogenicity of human breast cancer cells [2]. Therefore, BCL6 might play an important role in the development and progression of breast cancer.
MicroRNAs (miRNAs) are small, noncoding RNAs containing 18–24 nucleotides that negatively regulate gene expression by inhibiting target mRNA translation or by degrading target mRNA [3]. Recent studies have shown that miRNAs are useful candidate biomarkers for the early detection and prognosis of breast cancer. Most importantly, several studies have shown that miRNAs play an important role in regulating cell growth, differentiation, apoptosis, and carcinogenesis [45678]. In addition, we previously showed that BCL6 is the target gene of microRNA-339-5p (miR-339-5p) and that reduced miR-339-5p expression promotes BCL6 expression and alters the effects of BCL6 in breast cancer cells [29].
Prolactin (PRL) is a polypeptide hormone belonging to the growth hormone/cytokine family and is produced by autotrophs in the anterior pituitary gland and in normal and breast cancer tissues [10]. Tran et al. [11] showed that PRL-induced suppression of BCL6 expression was observed in xenotransplant tumors in nude mice in vivo and in freshly isolated human breast cancer explants ex vivo. However, previous studies have not provided novel evidences on whether PRL affects BCL6 expression or whether other PRL-induced pathways are involved in the suppression of BCL6 expression. Because miRNAs play an important role in tumor development and progression, we determined whether PRL prevented BCL6 activation in breast cancer cells through a miRNA-dependent pathway.
In the present study, we observed that PRL inhibited BCL6 mRNA and protein expression in breast cancer cells. Furthermore, miR-339-5p, a miRNA targeting BCL6, mediated PRL-induced BCL6 suppression, and blockade of miR-339-5p expression reversed the PRL-induced suppression of BCL6 expression. Ectopic stimulation with PRL inhibited the proliferation, colony formation, migration, and invasion of breast cancer cells. In contrast, suppression of miR-339-5p expression reversed the effects of PRL and promoted the proliferation, colony formation, migration, and invasion of breast cancer cells. Thus, we verified that the PRL–miR-339-5p–BCL6 pathway in breast cancer cells might be important for the development and progression of breast cancer.
METHODS
Reagents and antibodies
Antibodies against BCL6 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were obtained from Santa Cruz Biotechnology (Santa Cruz, USA). Cells were treated with 10 nM recombinant human PRL (ProSpec-Tany TechnoGene, Rehovot, Israel).
Cell culture
MCF-7, T47D, and SKBR3 human breast cancer cell lines were obtained from American Type Culture Collection (ATCC; Manassas, USA) and were cultured in ATCC-recommended conditions. The cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum at 37℃ in a humidified incubator with an atmosphere of 5% CO2.
Transfection of miRNA
For transient miRNA transfection, MCF-7 and T47D breast cancer cells were seeded (density, 1.0×106 per well) in 6-well plates and were grown overnight. On the next day, the cells were transfected with 2'-O-methylated single-stranded miR-339-5p antisense oligonucleotide (ASO; GenePharma, Shanghai, China) by using Lipofectamine 2000 (Invitrogen, Carlsbad, USA), according to the manufacturer's instructions. Negative control RNAs (GenePharma) was used to eliminate potential non-sequence-specific effects. The sequence of miR-339-5p ASO was 5'-CGUGAGCUCCUGGAGGACAGGGA-3' and those of non-sequence-specific negative control RNAs were 5'-UUCUCCGAACGUGUCACGUTT-3' (sense) and 5'-ACGUGACACGUUCGUAGAATT-3' (antisense). Transfection efficiency was confirmed by performing quantitative reverse transcription-polymerase chain reaction (qRT-PCR) and western blotting to determine miR-339-5p levels in the transfected breast cancer cells.
Protein extraction and Western blotting
Total cellular proteins were extracted and Western blotting was performed as described previously [1213]. Western blotting was performed using the following antibodies: rabbit anti-BCL6 polyclonal antibody (Santa Cruz Biotechnology) and mouse anti-GAPDH monoclonal antibody (Santa Cruz Biotechnology).
Cell proliferation and colony formation assays
After 48 hours of transfection, T47D cells were harvested and were subcultured in 96-well plates for up to 5 days. Cell proliferation was assessed by performing Cell Titer 96 AQueous MTS Assay (Promega, Fitchburg, USA), according to the manufacturer's instructions. Briefly, MTS reagent (20 µL) was added to each well, and the cells were incubated at 37℃ for 2 hours. Absorbance was measured at 570 nm by using a microtiter plate reader (Infinite M200; Tecan, Grödig, Austria). Next, the cells were cultured for 14 days, and colonies were counted. The experiment was performed in triplicate. Data are expressed as mean±standard deviation (SD).
Migration and invasion assays
Tumor cell migration and invasion were carried out using a Transwell insert (8 µm; Corning, New York, USA). In both the assays, the cells were seeded in a serum-free medium. The lower chamber of the transwell unit contained a medium supplemented with 10% serum that served as a chemoattractant. For the invasion assay, the inserts were precoated with extracellular Matrigel (BD Biosciences, Bedford, USA). The cells were incubated for 36 to 48 hours. Cells that did not migrate or invade through the pores were removed using a cotton swab. Filters were fixed using 90% ethanol, stained with 0.1% crystal violet, and photographed, and the number of cells was counted. Five low-magnification areas (×100) were randomly selected for counting the number of cells. Both the experiments were performed in triplicate.
Statistical analyses
All statistical analyses were performed using SPSS Software for Windows version 13.0 (SPSS Inc., Chicago, USA). Data are presented as mean±SD. Differences between groups were compared using Student t-test for continuous variables. The p-values less than 0.05 were considered statistically significant.
RESULTS
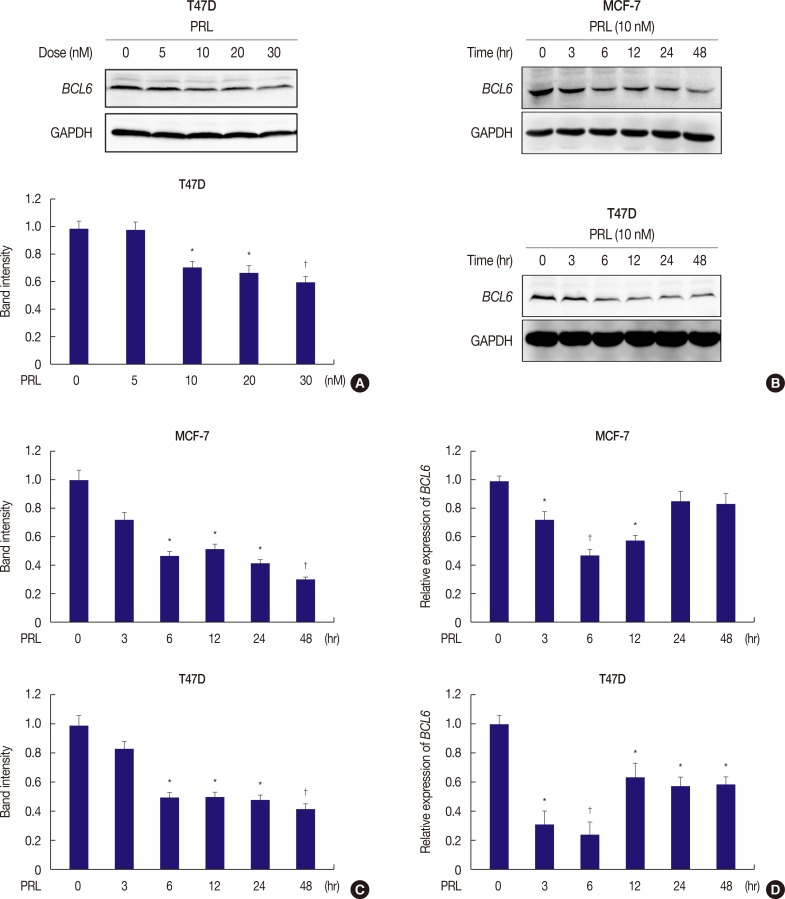
PRL suppresses BCL6 mRNA and protein expression in breast cancer cells
We determined BCL6 expression in response to treatment with different doses of PRL in T47D cells (Figure 1A) and later in MCF-7 and SKBR3 cells (Figure 1B, C, Supplementary Figure 1). Persistent treatment with 10 nM PRL for 6 hours markedly decreased BCL6 levels in MCF-7, T47D, and SKBR3 cells (Figure 1B, C, Supplementary Figure 1). Moreover, qRT-PCR, which was performed to determined BCL6 expression in MCF-7 and T47D cells, provided consistent results (Figure 1D). These results suggested that PRL negatively regulated BCL6 expression in human breast cancer cells.
PRL increases miR-339-5p expression in breast cancer cells
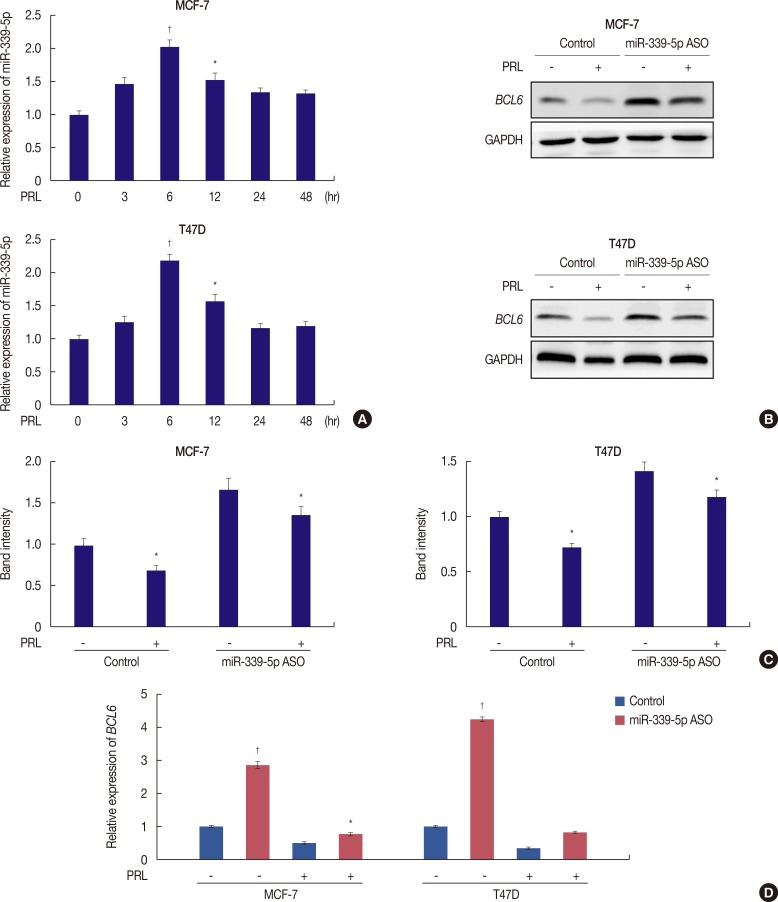
We next examined the involvement of miR-339-5p in PRL-mediated suppression of BCL6 expression. Results of qRT-PCR showed increased miR-339-5p expression in PRL-treated MCF-7 and T47D cells. This expression reached the highest level at 6 hours after PRL treatment, and was inversely associated with BCL6 mRNA expression (Figure 2A).
PRL regulates BCL6 expression through a miR-339-5p-dependent pathway
We next determined whether PRL regulated BCL6 expression through a miR-339-5p-dependent pathway. Quantitative RT-PCR and Western blotting were performed to determine the levels of BCL6 mRNA and protein expression in PRL-treated MCF-7 and T47D breast cancer cells. Downregulation of miR-339-5p expression by transfecting the specific ASO significantly increased BCL6 expression (Figure 2B, C). In addition, downregulation of miR-339-5p expression by transfecting MCF-7 and T47D cells with the specific ASO significantly abrogated the suppression of BCL6 mRNA and protein expression induced by treatment with PRL for 6 hours (Figure 2B, C). A similar result was obtained by performing qRT-PCR (Figure 2D). These results indicated that PRL downregulated BCL6 expression through a miR-339-5p-dependent pathway.
Inhibition of miR-339-5p expression alters the effects of PRL on breast cancer cells
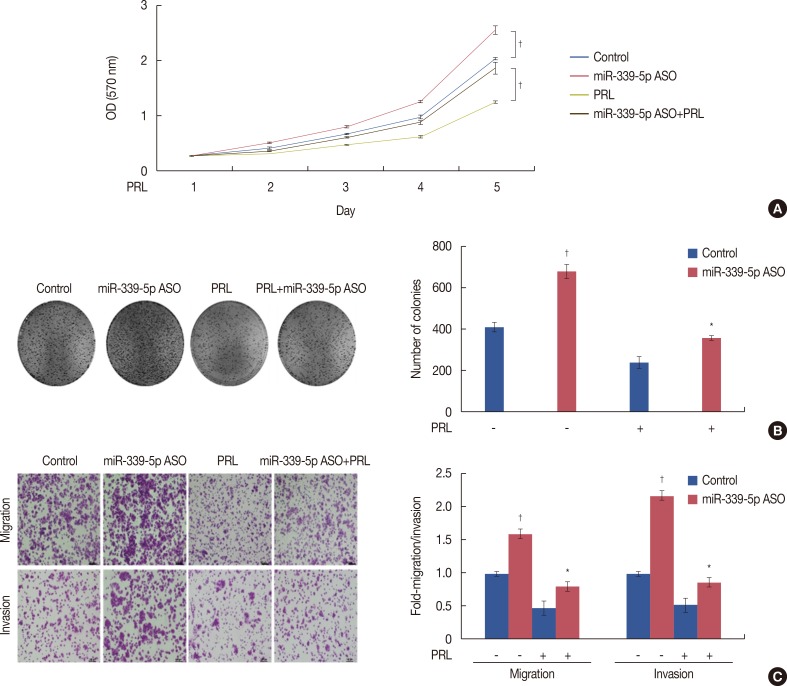
We examined whether the effect of PRL on the growth, migration, and invasion of breast cancer cells was mediated by miR-339-5p in vitro. Treatment with the miR-339-5p ASO alone significantly decreased cell viability. Next, we determined the effect of PRL on the growth of breast cancer cells. Analysis of cell viability indicated that PRL treatment for 6 hours decreased the proliferation of T47D cells compared with that of control cells. Suppression of miR-339-5p expression by transfecting the miR-339-5p ASO abrogated cell growth suppression induced by treatment with PRL for 6 hours (Figure 3A). To prevent toxicity induced by long-term drug exposure, cells were pretreated with the miR-339-5p ASO for 48 hours before stimulation with or without PRL for 6 hours. Similar results were obtained in the colony formation assay (Figure 3B).
We next determined the potential impact of PRL on the migration and invasion capacity of breast cancer cells. Because T47D cells showed weak migration and invasion capacity, we used MCF-7 cells to assess the effect of PRL on the migration and invasion capacity of breast cancer cells. The transwell assay showed that miR-339-5p knockdown increased the migration and invasion rate of MCF-7 cells and that PRL treatment significantly decreased the migration and invasion capacity of MCF-7 cells compared with that of control cells (Figure 3C). However, downregulation of miR-339-5p expression by transfecting the ASO in the presence of exogenous PRL stimulation significantly increased the migration and invasion capacity of MCF-7 cells compared with that of cells treated with only PRL (Figure 3C).
DISCUSSION
The present study provides an evidence of a novel pathway underlying PRL-induced suppression of BCL6 expression in breast cancer cells. PRL-treated MCF-7 and T47D cells showed high levels of miR-339-5p expression and markedly decreased levels of BCL6 mRNA and protein expression. Furthermore, PRL-induced inhibition of BCL6 expression was re-versed by the ASO-mediated suppression of miR-339-5p expression. Growth of T47D cells was reduced after exogenous
PRL stimulation, and suppression of miR-339-5p expression reversed the effects of PRL on the proliferation, colony formation, migration, and invasion of breast cancer cells. These results indicated that miR-339-5p played an important role in the PRL-BCL6 signaling pathway during the progression of breast cancer.
Our previous study showed that BCL6 expression was regul-ated by miR-339-5p and that BCL6 was the direct target of miR-339-5p [2]. Upregulation of BCL6 expression may exacerbate the biological consequences associated with the loss of miR-339-5p signaling in breast cancer cells because of the suppressive effect of BCL6 on miR-339-5p target gene induction [2]. A strong negative correlation between BCL6 expression and miR-339-5p in normal and malignant breast tissues supports the selective role of miR-339-5p as a suppressor of BCL6 expression, as suggested by the in vitro data obtained in our study and those of previous studies [29].
PRL, a hormone secreted from the anterior pituitary gland, is involved in the growth and differentiation of breast epithelia during pregnancy and lactation [14] and in the initiation and progression of breast cancer [1015]. Effects of PRL are mediated by its receptor (PRLR). Binding of PRL to PRLR initiates signaling cascades through multiple downstream partners, including Janus kinase 2 (JAK2) [1016]. Most physiological effects of PRL on the mammary gland are mediated by JAK2/signal transducer and activator of transcription 5 (STAT5) pathway [17]. In breast cancer cells, activation of STAT5 predicts favorable clinical outcomes [18]. Tran et al. [11] reported that PRL inhibits BCL6 expression in breast cancer cells through a STAT5a-dependent pathway. Because the consensus DNA-binding sequence of BCL6 resembles that of STAT5, BCL6 competes with STAT5 for binding to various interaction sites on DNA [192021]. Sato et al. [22] suggested that PRL suppressed progestin-induced BCL6 expression through the JAK2/STAT5 pathway. However, to the best of our knowledge, a crosstalk between PRL-induced pathway and miRNAs has not been reported to date. Previous studies have suggested that BCL6 is regulated by miRNAs because it contains miRNA-binding sites in its 3'-untranslated region [4]. We hypothesized that miRNAs are involved in the PRL-induced BCL6 suppression in breast cancer cells. High circulating levels of PRL are a risk factor for metastatic ERα+ breast cancer [2324]. MCF-7 and T47D cell lines are 2 of the most studied ERα+/PRLR+ luminal breast cancer cell lines to determine the effects of estrogen and PRL [252627]. Therefore, we chose these two cell lines to verify whether miRNAs mediate PRL-induced suppression of BCL6 expression and to determine the effects of miRNA and PRL crosstalk on the growth, proliferation, migration, and invasion of breast cancer cells.
A previous study showed that miR-339-5p regulates the proliferation, migration, and invasion of different cancer cells and that downregulation of miR-339-5p expression increases the migration and invasion of breast cancer cells [2,9,28]. Therefore, we hypothesized that PRL enhanced miR-339-5p expression, which in turn suppressed BCL6 expression. In the present study, we used an in vitro model system to prove our hypothesis. First, we observed that the growth of T47D cells was reduced upon exogenous PRL stimulation. Consistent with this result, Nitze et al. [29] suggested that autocrine PRL signaling was a general mechanism that promoted tumor growth in patients with breast cancer and that ectopic PRL reduced the growth of T47D cells. Another study identified PRL as a critical regulator of epithelial plasticity and a suppressor of invasion of breast cancer cells [30]. In the present study, we observed that transfection of miR-339-5p ASO significantly abrogated the suppressive effect of PRL on the proliferation, colony formation, migration, and invasion of breast cancer cells.
In summary, the present study identified a novel PRL–miR-339-5p–BCL6 pathway involved in the regulation of human breast cancer progression. To the best of our knowledge, this is the first study to show the crosstalk between miRNAs and PRL in breast cancer. Multiple factors and signaling pathways may be involved in the PRL-induced regulation of breast cancer progression, of which miRNA signaling appears to be an important pathway. However, future studies are required to elucidate mechanisms underlying PRL-induced regulation of miRNA expression.

 XML Download
XML Download