

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cancer is a polygenic disease of the eukaryotic cell cycle, involving genetic and epigenetic alterations in the genes playing a vital role in controlling the cell cycle [1]. Cell cycle progression is a highly organized series of events, involving multiple checkpoints to monitor growth signals and DNA integrity during cell division [2]. Any dysregulation at these checkpoints due to aberrant expression of the genes involved in the cell cycle machinery (E2F5/pRb) may lead to proliferation and carcinogenesis [3]. Among these events, G1/S transition is crucial for maintaining DNA conservation and this step is actively controlled by the availability of E2F family of transition factors [4]. E2F is a protein family comprising nine members, four of which are transcriptional activators (E2F1–E2F3) and five are transcriptional repressors (E2F4–E2F8) [5]. The E2F family can bind with Rb proteins and regulate the G1/S phase of cell cycle [6]. These cell cycle regulatory genes, along with the associated regulatory factors, play a fundamental role in the proliferation of cells and pathogenesis of cancer [7]. Overexpression of the activator E2Fs and downregulation of the repressor E2Fs may cause abnormal proliferation of cells, a common phenomenon in various types of malignancies [3].
Among the E2F family, the E2F5 gene has growth-repressive characteristics that have been observed in solid osteosarcoma [8] and cancer of the colon [9], ovary [3], and breast [10]. Overexpression of E2F5 proteins has been observed in both ovarian and breast cancers [310]. Moreover, elevated expression of E2F5 has been reported in early as well as advanced stages of ovarian cancer, which suggests a putative role of E2F5 in cancer pathogenesis [3]. These elevated levels of expression have been attributed to many genetic factors [11] such as abnormal splicing causing impaired DNA amplicons in sporadic colorectal carcinoma [9] and chromosomal aberrations involving the E2F5 gene in breast cancer [10]. However, the role of epigenetic modifications such as DNA methylation in the regulation of E2F5 remains largely unclear.
Many epigenetic alterations such as DNA and histone methylation as well as chromatin remodeling functions such as histone acetylation, phosphorylation, and ubiquitination are crucial for regulating the growth patterns of many genes [12]. Among these, promoter methylation at the CpG sites is the most important epigenetic tag that may correlate with gene expression [13]. Moreover, CpG methylation at the promoter region of a gene may correspond to the gain of function of oncogenes and loss of function of the tumor suppressor gene, thus contributing key characteristics that can lead to tumorigenesis [14]. Aberrant DNA methylation has been observed in tumor cells and is thought to be helpful to develop disease markers for early prognosis of cancer pathogenesis [1516].
This study attempts to elucidate E2F5 promoter methylation in breast cancer tissue to highlight a possible role in cancer pathogenesis, which may be used for early prognosis of breast cancer in clinical settings.
METHODS
Subject enrollment and sample collection
Fifty tumor tissue samples and adjacent normal control tissue (ANCT) samples were collected from breast cancer patients in RNAlater® stabilization solution (Thermo Fisher, Waltham, USA) at the time of surgery at Khyber Pakhtunkhwa. ANCT was selected from the area about 2 cm away from the affected site and selected by an oncologist on the basis of histopathological observations. Patients with history of other infectious and familial diseases were excluded from this study. This study was conducted after prior approval from the Ethical Review Committee (CIIT-09-10-14) of the COMSTAS Institute of Information Technology (CIIT), Islamabad, Pakistan, and the collaborating hospital. Written informed consent was obtained from every patient, prior to sample collection. Samples were stored at -80℃ until analysis.
Extraction and bisulfite conversion of genomic DNA
DNA from the tissue samples was extracted using the standard phenol-chloroform method [17]. About 2 to 5 µg of genomic DNA was chemically modified using the commercially available EpiJET™ Bisulfite Conversion Kit (Cat# K1461; Thermo Scientific, Waltham, USA) as per the manufacturer's instructions. Bisulfite treatment specifically converts nonmethylated cytosine "C" residues to thymine "T" residues, but it retains methylated cytosine "C."
Primer designing for methylation-specific polymerase chain reaction (PCR)
E2F5 methylation status was analyzed in both sets of tissues. The targeted region is located at a site 523 bp downstream from the transcription start site. Moreover, methylation-specific primers were designed for the targeted site (NCBI "85177579-85178045") and comprised 51 CpG sites. Promoter methylation was analyzed using a set of methylated (forward: TGGGGTTGTTTATTATTAAGTTCGT and reverse: ACGATATCTCCGAACAAAACG) and nonmethylated primers (forward: GAGTTTGGGGTTGTTTATTATTAAGTTT and reverse: CACAATATCTCCAAACAAAACACC) on the MethPrimer freely online available tool (http://www.urogene.org/methprimer/) [18].
Methylation-specific PCR
Methylation-specific PCR (MSP) was performed to amplify the targeted promoter region of E2F5 from the bisulfite-converted genomic DNA by using methylation-specific primers. Briefly, 4 µL (~100 ng) bisulfite-converted genomic DNA was amplified using methylated and nonmethylated primers in a 50-µL reaction mixture. The PCR conditions were as follows: a single cycle at 98℃ for 30 seconds (initial denaturation); followed by 35 cycles at 98℃ for 10 seconds (denaturation), 62℃ for 30 seconds (annealing), and 72℃ for 30 seconds (extension); and a single cycle at 72℃ for 7 minutes (final extension) by using 1.0 units Thermo Scientific™ Maxima Hot Start Taq DNA Polymerase™ (Cat# EP0601; Thermo Scientific). MSP validity was checked by amplifying CpG-methylated human genomic DNA obtained from Thermo Scientific Cat# SD1131 (Thermo Scientific) as the positive control; PCR water was used as the negative control. However, for the nonmethylation-specific PCR, bisulfite-unconverted human genomic DNA was used as the positive control.
Gel electrophoresis and quantification
PCR products were visualized under the UV illuminator BioDoc Analyze™ (Biometra, Göttingen, Germany) after electrophoresis on 2% agarose gel, followed by staining with ethidium bromide. The bands intensities were quantified using ImageJ densitometer, freely available online (Laboratory for Optical and Computational Instrumentation, University of Wisconsin, Madison, USA) to compare the degree of methylation and nonmethylation. Change in methylation status (Δmeth) was calculated by subtracting the intensity of methylation from that of nonmethylation. CpG human methylated DNA was used as the positive control, while the nonmethylated material was used as the control.
mRNA extraction and real-time qPCR analysis
RNA extraction from all tissues was performed using the standard Trizol method [19], and cDNA was synthesized using the commercially available cDNA synthesis kit (Thermo Scientific). Following were the primers used: E2F5 gene (left primer, CACCTTCTGGTACACAACTG; right primer, GGCTTAGATGAACTCGACTC); β-actin as the internal control (left primer, CACTCTTCCAGCCTTCCTTC; right primer, TGATCTCCTTCTGCATCGTG). StepOnePlus™ Real-Time PCR system (Applied Biosystems, Waltham, USA) was used for PCR amplification by the SYBR® Green method. The thermocycler was set to the following conditions: 95℃ for 10 minutes (initial denaturation); followed by 40 cycles at 95℃ for 15 seconds, 56℃ for 60 seconds, and 72℃ for 20 seconds; and final extension at 72℃ for 1 minute. mRNA expression was calculated using the delta-delta Ct values.
Data analysis
Data were analyzed using OriginPro 2015 statistic software (OriginLab, Northampton, USA). The correlation among different factors was assessed at 95% confidence intervals by using the Mann-Whitney test, Kruskal Wallis test, and ANOVA test, and specific comparison were undertaken by standard descriptive analysis. A p-value of <0.05 was considered significant.
RESULTS
E2F5 promoter methylation analysis among tumor and normal tissues from breast cancer patients
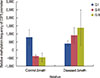
The methylation status of E2F5 promoter was analyzed for both tumor tissues and ANCTs in breast cancer. Both methylation- and nonmethylation-specific primers yielded products in case of normal tissue, whereas majority of tumor tissues yielded products only with nonmethylation-specific primers (Figure 1). Reduced methylation level at the E2F5 promoter region (48%) was observed in the tumor tissues compared to the normal tissues (80%), as shown in Table 1 and Figure 2. Δmeth was found to be statistically different (p=0.022) between normal and tumor tissues (Table 2). This highlights a possible role of E2F5 promoter methylation in breast cancer prognosis, which can be correlated to its well-reported elevated expression levels in many cancer types.
Promoter methylation status among patients from different age groups
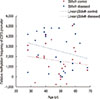
The promoter methylation status of the E2F5 gene was studied between two study cohorts of breast cancer, namely, <45 and ≥45 years. Interestingly, the methylation frequency was higher among control tissues, compared to the tumor tissues, among our subjects. In both study cohorts, the methylation frequency in the control tissues was higher (63.6% and 92.9%, respectively), compared to that in the tumor tissues (27.3% and 64.3%, respectively). Statistical analysis showed that changes in the methylation status (Δmeth) are significantly related to both tissue types (p<0.001) (Tables 1, 2, Figure 3). These results propose the occurrence of age-independent changes in the E2F5 promoter methylation status, which points toward a more direct involvement of methylation in breast cancer.
Promoter methylation analysis among premenopausal and postmenopausal patients
Methylation frequencies were also analyzed among premenopausal and postmenopausal breast cancer patients. Of the total subjects (n=25), 13 belonged to the premenopausal group and 12 patients belonged to the postmenopausal group. The methylation frequency in premenopausal patients was lower in the tumor tissues (23.1%); it was 75.0% in postmenopausal patients. Moreover, in both cohorts, the frequency of methylation was higher in the control group. Moreover, the methylation levels were significantly different (p<0.001) among patients of the premenopausal and postmenopausal groups (Tables 1, 2).
Methylation frequency among different stages and grades of breast cancer
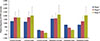
Breast cancer patients with different clinical manifestations or at different stages were analyzed for their E2F5 promoter methylation status. In patients at initial stages of tumor progression (i.e., Stage I), the methylation frequency was lower (43.8%) compared to patients at an advanced stage (Stage II, 50%; Stage III, 66.7%). This suggests a potential role of E2F5 promoter methylation in disease progression, and highlights its prognostic value as an early biomarker. Δmeth was significantly different (p<0.001) among patients of all disease groups (Tables 1, 2, Figure 4). Similar results were observed in patients with Grade I (33.3%), Grade II (42.1%), and Grade III (100%) disease. Δmeth was also found to be significantly different (p<0.001) among the three tumor classes (Tables 1, 2, Figure 5). This indicates potential involvement of E2F5 methylation in disease progression. Moreover, Δmeth is a progressive epigenetic factor that could significantly modulate tumor biology at a more advanced disease stage in the sample set (Pearson's r=0.99) (Table 3).
Methylation status among various histopathological groups of breast cancer
Methylation levels among various histopathological classes of breast cancer were also analyzed. Among the three histopathological classes, patients with invasive ductal carcinoma had 46.7% methylation in the tumor tissues compared to those with invasive lobular carcinoma (80%). Moreover, patients with ductal carcinoma in situ showed 20% methylation in tumor tissues. All three classes showed a significant difference (p<0.001) in their methylation status (Tables 1, 2, Figure 6). This suggests that the methylation level of the E2F5 promoter might have varying consequences depending on the origin of the tumor.
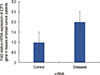
E2F5 mRNA transcript expression is significantly upregulated in tumor tissues
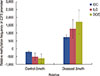
Quantitative real-time PCR was performed to determine E2F5 mRNA transcript expression in both tumor and control tissues. The relative expression of E2F5 mRNA transcript was found to be significantly higher (p<0.001) in the tumor tissues compared to the ANCTs, irrespective of the age and disease status of the patient (Table 2, Figure 7). Moreover, Pearson's correlation among the relative expression in the control and tumor samples with their Δmeth shows negative correlation (-0.09 and -0.22, respectively), which suggests the possible role of E2F5 promoter methylation in its upregulation, leading to breast cancer pathogenesis (Table 4).
DISCUSSION
Cell cycle regulatory genes and associated regulatory proteins are vital factors that play a critical role in cell proliferation and tumorigenesis. Promoter methylation has a critical role in the regulation of gene expression, resulting in normal growth pattern [13]. E2F5 gene is cytogenetically mapped to the human chromosome 8q21.2 and comprises 10 exons [20]. They have a highly conserved DNA-binding domain, Dp-1 domain for dimerization, and a transactivation domain in the C-terminus of the protein. The DNA-binding domain of E2F5, in association with DP-1, binds with the target promoter and regulates the expression of the reporter gene [21]. Unlike E2F1–3, E2F4 and E2F5 are highly expressed in quiescent (G0) cells, and associate with retinoblastoma-related proteins (p107/p130), instead of pRB, and show antioncogenic activity [21]. Both Rb1/p107 and Rb2/p130 form complexes with E2F4 or E2F5, resulting in the formation of cyclin E repressor complex (CERC) [22]. These CERC play a pivotal role in the transcriptional repression of Cyclin E1 promoter in the G1 phase and in the blockade of cell proliferation. Thus, aberrant expression of the E2F5 gene may lead to uncontrolled cell division and tumor formation. Higher E2F5 expression has already been reported in various tumor types, including colon [9], ovarian [3], and breast cancer [10], but the factors regulating E2F5 gene expression remains elusive. Moreover, cBioportal showed significantly high expression of E2F5 gene in 281 (26%) breast cancer patients out of 1,100 patients, although E2F5 expression profiles were not significantly correlated with patient survival data on the same portal [23]. Our results also support the observation that higher E2F5 expression occurs in malignant tissues. Moreover, our results indicate that E2F5 is aberrantly methylated to a lesser extent in the breast tumor tissues (48% of all cases), compared to the control samples (80%). ΔMeth status was found to be significantly different (p=0.022) among the control and tumor tissues. Deregulated methylation at the E2F5 gene promoter may be considered responsible for elevated expression levels in breast tumor tissues and may have a prognostic role; however, this needs further validation in other tumor types. Alternatively, higher methylation levels at the E2F5 promoter may extend protective effects against breast cancer pathogenesis. This is supported by the fact that lower methylation frequencies were noticed for breast tumor tissues in all control study cohorts (age-based, tumor stage-based, and so forth). Similarly, a gradual decrease in methylation levels was observed at the E2F5 promoter in the breast cancer patients at an advanced stage. This observation highlights the prognostic involvement of E2F5 promoter in tumor progression and can be used as an early diagnostic marker of breast cancer. This is supported by the fact that well-differentiated (Grade I) tumors are less methylated compared to poorly differentiated tumors (Grade III). This might furtehr detrioate other cellular mechanisms leading to complications at this disease stage that have adverse effects on disease management, owing to poor socioeconmic sittuation of Pakistani population [24]. Interestingly, tumors of different tissue origins (histopathological groups) were found to have varying degrees of methylation levels, suggesting that the consequences of E2F5 promoter methylation might have different effects depending on the area of tumor origin. The level of methylation increased as the tumors progressed to the advanced stages and showed significantly reduced expression of the tumor suppressor gene [25].
Generally, promoter methylation is considered an age-dependent phenomenon [26]. It is hypothesized that age-dependent DNA hyper-methylation is associated with aberrant telomerase activities and plays a critical role in age-related disorders such as cancer [27]. The present study shows an ageindependent change in methylation status at the E2F5 gene promoter. In both study cohorts (<45 and ≥45 years), the difference in methylation was negligible, given the fact that the methylation status was statistically different from that of the corresponding controls (p<0.01) (Table 2). This suggests a more direct involvement of E2F5 promoter methylation in breast cancer pathogenesis and supports our aforementioned hypothesis that E2F5 promoter methylation does exert a protective effect against breast cancer.
Many studies have reported C/T transitions in the coding and regulatory regions of the genes involved in aging and various cancers and related disorders [28]. Consistent with this is the fact that the promoter-rich CpG sites are hotspots for methylation and that they are vulnerable to mutagens [29]. The conversion of cytosine to thymidine renders methylation at that position, which may induce pronounced effects on gene expression [30]. Interestingly, the E2F5 gene has 27 reported C/T conversions in the chromosomal region (8q21.2) >gi|568815590:85177579-85178045 present about 523 bp around the E2F5 TSS. This supports our findings that methylation at the E2F5 promoter has a prognostic role in breast cancer, although no literature is available on disease susceptibility for the population harboring these single nucleotide polymorphisms. Based on these findings, screening C >T conversion in normal individuals seems to be an interesting domain to explore.






 XML Download
XML Download