

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Trabectedin is a natural marine compound isolated from the Caribbean tunicate Ecteinascidia turbinata [1]. Trabectedin is currently used for the treatment of patients with soft tissue sarcomas after failure of anthracyclines and ifosfamide, or for whom these drugs are unsuitable. It is also used in combination with pegylated liposomal doxorubicin for the treatment of patients with relapsed, platinum-sensitive ovarian cancer [23]. Trabectedin shows a unique mechanism of action, as the drug is able to interact with proteins involved in DNA repair, in addition to inhibiting activated transcription [4567]. For example, it was proposed that trabectedin adducts trap members of the nucleotide-excision repair (NER) system (e.g., XPG), forming large ternary complexes [68]. These not only inhibit NER activity, but also stimulate cleavage by the XPF/ERCC1 nuclease on the strand opposite to that bonded by the drug, generating single strand breaks (SSBs) [679]. Ternary complexes or SSBs generated by XPF/ERCC1 stall replication forks, leading to double-strand DNA breaks (DSBs). As evidence of this, it has been demonstrated that cell lines deficient in different NER proteins were less sensitive to trabectedin [6810]. Conversely, defects in homologous recombination (HR) were associated with higher sensitivity to the drug, indicating that trabectedin can induce synthetic lethality [1011].
Synthetic lethality represents a new paradigm for cancer treatment [12]. This concept describes a genetic interaction in which single-gene defects are compatible with cell viability, but the combination (or "synthesis") of various gene defects results in cell death [13]. Synthetic lethality provides a potential mechanistic framework for the therapeutic targeting of genetic and functional deficiencies in cancers and is currently under investigation. For example, inhibition of poly-(adenosine diphosphate ribose)-polymerases (PARPs) has been shown to enhance platinum sensitivity in breast and ovarian cancer models carrying mutations in BRCA1 and BRCA2 genes [14]. This is due to the accumulation of more lethal DSBs in a context of HR deficiency after treatment with this drug combination [15]. In this manuscript, we have hypothesized that the combination of trabectedin with a PARP inhibitor could be a useful strategy to treat breast tumors. Theoretically, combining NER inhibition by trabectedin with PARP inhibition could create artificial synthetic lethality resulting in a synergistic antitumor effect. To demonstrate this hypothesis, (1) we have investigated drug synergism in vitro for combinations of trabectedin and the PARP inhibitors veliparib, olaparib, and iniparib in different proficient- or deficient-BRCA1 breast tumor cells; and (2) we have evaluated the cellular and molecular effects induced by combinations versus single agents by comparing treatment-induced DNA damage, protein poly(ADP-ribosyl) ation (PARylation), and cell cycle perturbations.
METHODS
Reagents
Trabectedin (Yondelis®) was manufactured by PharmaMar S.A. (Madrid, Spain). Veliparib, olaparib, and iniparib were purchased from Selleck Chemicals (Munich, Germany). Stock solutions of drugs were prepared in pure DMSO at the appropriate concentrations and stored at -20℃ until use. Propidium iodide (PI), 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and antibodies against α-tubulin (T5168) were obtained from Sigma (St. Louis, USA). Antibodies against FEN1 (ab17993), DNA Pol β (ab26343), XRCC1 (ab1838), FANCD2 (ab2187), and ATM (Y170) were obtained from Abcam (Cambridge, UK). Antibodies against DNA-PK (#4602) and BRCA1 (#9010) were obtained from Cell Signaling Technologies (Danvers, USA). Antibodies against PARP-1 (sc-7150) and XPD (sc-20696) were obtained from Santa Cruz Biotechnology (Santa Cruz, USA). Antibodies against γ-H2AX (05-636), PAR (#551813), XPF (MS-1381), and XPG (A301-484A) were obtained from Merck Millipore (Billerica, USA), BD Pharmigen (San Jose, USA), NeoMarkers (Fremont, USA), and Bethyl Lab (Montgomery, USA), respectively.
Cell lines
Breast tumor cell lines MCF7 (ATCC HTB-22), MDA-MB-231 (ATCC HTB-26), MDA-MB-436 (ATCC HTB-130), and HCC-1937 (ATCC CRL-2336) were obtained from the American Type Culture Collection (ATCC, Rockville, USA). MCF7 tumor cells do not present mutations and are thus considered as BRCA1+/+ (Table 1) [16]. MDA-MB-231 tumor cells present a heterozygous mutation in the BRCA1 gene and are considered BRCA1+/- cells [16]. MDA-MB-436 and HCC-1937 tumor cells present a homozygous mutation in the BRCA1 gene and are considered BRCA1-/- cell lines [16]. Cells were maintained at 37℃ and 5% CO2 in Dulbecco's modified eagle medium (DMEM) supplemented with 10% fetal-bovine serum (FBS), 2 mM L-glutamine, and 100 units/mL of penicillin-streptomycin.
Clonogenic assays
For clonogenic assays, tumor cells were seeded on six-well plates at a density of 2,000 cells/well and incubated with the appropriate concentration of each drug. After 120 hours, cells were harvested, fixed with 0.5% glutaraldehyde, and stained with sulforhodamine B. Colonies were then counted in at least two cultures. Concentration-response curves and their respective IC50 values (concentration of drug that produces a 50% inhibition of cell growth) were calculated using Prism v5.02 software (GraphPad, La Jolla, USA). Experiments were performed in duplicate.
Combination studies
Combined effects of trabectedin and PARP inhibitors were determined using MTT assays. Briefly, all cell lines were seeded in 96-well microtiter plates at the appropriate cell density and treated with vehicle, each drug alone, or combinations of trabectedin and PARP inhibitors for 72 hours. Then, MTT was added to each well and its absorbance measured at an optical density of 540 nm with a POLARStar Omega Reader (BMG Labtech, Offenburg, Germany). IC50 values were obtained by iterative nonlinear curve fitting using Prism 5.02 statistical software (GraphPad, La Jolla, USA). The data presented are the average of at least three independent experiments performed in triplicate. Combinations of trabectedin and PARP inhibitors were evaluated using a nonconstant ratio design. The following standard potency ratios (%IC50trabectedin/%IC50PARP Inh) were used: 50/50 or equipotent ratio, 40/60, 60/40, and 75/25. For each potency ratio, we have calculated their equivalent concentration ratios (n-fold concentration of the PARP inhibitors with respect to trabectedin).
Data were analyzed for synergistic effects using the median-effect method. Synergism was defined as a greater-than-expected additive effect while antagonism is defined as less-than-an-expected additive effect. Thus, combination index (CI)=1 indicated an additive effect, CI <1 indicated a synergistic effect, and CI >1 indicated antagonism. Because CI values may change with the fraction affected in a nonlinear manner, the CI should optimally be presented for each effective dose (ED) tested with valid results. In our experiments, effective concentrations were ED20, ED50, and ED80 and indicated the compound concentration that resulted in 20%, 50%, and 80% cell death, respectively. The CI values obtained for the combination studies were calculated using CalcuSyn software (Biosoft, Cambridge, UK).
Western blot assays
Tumor cells were treated with the appropriate concentration of each compound or combination at different time intervals and resuspended in lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% (v:v) Nonidet P-40, 2 mM EDTA, 1 mM PMSF, 10 µg/mL aprotinin, and 10 µg/mL leupeptin) and kept on ice for 15 minutes. Protein content was determined using the modified Bradford method. Cellular proteins were fractionated on SDS-PAGE and then transferred onto PVDF membranes (Immobilon-P; Millipore, Billerica, USA). Membranes were subsequently incubated with the appropriate primary antibody for 24 hours, then washed and incubated with the corresponding secondary antibody. Finally, proteins were visualized using an ECL System (GE Healthcare, Fairfield, USA).
Comet assay
For the comet assay, a single-cell gel electrophoresis assay was used (Trevigen, Gaithersburg, USA). Briefly, all cell lines were treated with the appropriate drug for 6 hours (MDA-MB-231) or 12 hours (MCF7, MDA-MB-436, and HCC-1937); then, cells were detached mechanically and added to low-melting-point agarose. After lysis, cells were subjected to electrophoresis and the comets were stained with SYBR-Green. Pictures were taken with a Leica DM IRM fluorescence microscope equipped with a DFC 340 FX digital camera (Leica, Wetzlar, Germany). Quantitation of the Tail Moment (a damage measure combining the amount of DNA in the tail with the distance of migration) was performed with CometScore software (TriTek, Sumerduck, USA). For each condition, a minimum of 50 cells were analyzed. The experiment was performed in duplicate.
Cell cycle analysis
Tumor cells were treated with the appropriate amount of each compound or combination for 24 hours, fixed in 70% ice-cold ethanol for 12 hours at -20℃, treated with RNase A (100 µg/mL), and then stained with 50 µg/mL PI for 30 minutes at 37℃. Samples were acquired and analyzed with a BD Accury C6 flow cytometer (Beckton and Dickinson, Franklin Lakes, USA) and the Modfit LT 4.1 software (Verity Software, Topsham, USA), respectively.
RESULTS
Antiproliferative activity of trabectedin and PARP inhibitors in breast tumor cell lines
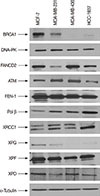
We measured the levels of expression of BRCA1 as well as other proteins involved in DNA repair systems (DNA-PK, FANCD2, ATM, FEN1, DNA polymerase β, XRCC1, XPF, and XPD) in the four breast cancer cell lines by western blot (Figure 1). As expected, BRCA1 was detected only in MCF7 and MDA-MB-231 cells. HCC-1937 cells expressed low levels of a truncated form of BRCA-1 protein, as evidenced by a higher electrophoretic mobility than the wild type protein. Interestingly, these cells also showed higher levels of DNA polymerase β and XRCC1 compared to the other three cell lines. Another marked difference among the cell lines was the expression of XPG. This NER-related endonuclease was detected in MCF7 and MDA-MB-231 cells, but not observed in MDA-MB-436 or HCC-1937 cells. Finally, FANCD2 expression was down-regulated in MDA-MB-231 cells. No other major differences were observed among the breast cancer cell lines.
We analyzed the antiproliferative activity of trabectedin and the three PARP inhibitors in a clonogenic assay. The IC50 values for trabectedin, veliparib, olaparib, and iniparib are shown in Table 2. As expected, BRCA1-deficient MDA-MB-436 cells were more sensitive to PARP inhibitors (particularly veliparib and olaparib) compared to MCF7 and MDA-MB-231 cells. HCC-1937 cells were less sensitive to veliparib and olaparib, which could be due to the overexpression of DNA polymerase β and XRCC1. Of note, all the cell lines were highly sensitive to trabectedin, despite MCF7 and MDA-MB-231 cells expressing high levels of BRCA1, which may be due to high levels of XPG.
Combination studies
The combination of trabectedin with each of the three PARP inhibitors was analyzed using a nonconstant ratio design in which the two drugs were combined in different ratios for each combination. Synergistic antiproliferative effects (CI<1) were only observed when treating the four breast cancer cell lines with the combination of trabectedin and olaparib (Table 3). In MCF7 cells, the combination resulted in CI values that ranged from 0.27 (ED80 at 1:3,000 trabectedin:olaparib concentration ratio) to 0.9 (ED20 at 1:26,600 trabectedin: olaparib concentration ratio). In MDA-MB-231, the combination resulted in CI values that ranged from 0.34 (ED80 at 1:101,600 trabectedin:olaparib concentration ratio) to 0.81 (ED20 at 1:5,000 trabectedin:olaparib concentration ratio). In MDA-MB-436 cells, the combination resulted in CI values that ranged from 0.33 (ED50 at 1:126,600 and 1:84,800 trabectedin:olaparib concentration ratios) to 0.65 (ED80 at 1:26,600 trabectedin:olaparib concentration ratio). In HCC-1937 cells, the combination resulted in CI values that ranged from 0.65 (ED20 at 1:10,000 trabectedin:olaparib concentration ratio) to 0.80 (ED80 at 1:10,000 trabectedin:olaparib concentration ratio). Synergistic effects were not observed when trabectedin was combined with veliparib or iniparib. Overall, the trabectedin/olaparib combination showed a strong synergistic effect on human breast tumor cells, regardless of BRCA1 status. Since the combinations of trabectedin with veliparib or iniparib did not exhibit synergy, further studies were performed only with the trabectedin/olaparib combination.
Analysis of cell cycle perturbations and apoptosis
Figure 2 summarizes the results of cell cycle perturbations after 24 hours of exposure to trabectedin and olaparib alone and in combination in the four breast cancer cell lines. In MCF7, MDA-MB-231, and HCC-1937, the combination induced a decrease of the G0/G1 peak with an accumulation of cells in the G2/M peak. This effect was stronger with the combination than the activity of either compound used as a single agent. Of note, MDA-MB-436 was revealed to be an aneuploidy cell line (diploid population, 34.6%; aneuploidy population, 65.4%); the combination induced mostly an S-phase arrest in the diploid subpopulation.
Analysis of PARylation and DNA damage
PARylation significantly triggers the accumulation of several DNA damage response (DDR) proteins at the DNA lesions and is therefore a marker of DNA damage. We evaluated the effects of trabectedin, olaparib, and combination in PAR synthesis after 72 hours of incubation to determine the extent of this effect. Treatment of breast cancer cells with trabectedin induced PAR expression at the highest concentration tested (Figure 3). Conversely, olaparib reduced PAR expression in all the cell lines as expected (Figure 3). The combination of trabectedin and olaparib reduced the expression of PAR; however, the extent of inhibition was lower than that observed with olaparib alone.
To quantify the extent of DNA damage, we also analyzed the γ-H2AX expression after the different drug treatments using western blot. As shown in Figure 3, the combination of trabectedin and olaparib induced higher levels of γ-H2AX expression in all breast cancer cell lines. The expression of γ-H2AX was evident even with concentrations of trabectedin as a single agent that did not induce the formation of DSB. The induction of DNA strand breaks was further evaluated using a comet assay. In this assay, DNA from cells with accumulated damage appears as fluorescent comets with tails of fragmented or unwound DNA, whereas normal, undamaged DNA does not migrate far from the origin (Figure 4). We observed a clear concentration-dependent increase in DNA strand breaks in all cell lines following treatment with the combination (Figure 4). Altogether, these results are consistent with the synergy observed in the antiproliferative studies.
DISCUSSION
The purpose of this study was to evaluate the effect of trabectedin when combined with a PARP inhibitor (veliparib, olaparib, or iniparib) in a panel of human breast cancer cell lines, including cell lines with BRCA1-mutation. Our results show a synergistic effect only for the combination of trabectedin and olaparib. This combination induced G2/M arrest and caused higher levels of DNA damage than each drug alone. The synergistic effects were independent of the BRCA1 status of tumor cells in the cell models selected for this study. Synergy was not observed with the other two PARP inhibitors used in the study. Altogether, our data suggest that combination treatment with trabectedin and olaparib may be more effective than monotherapy in treating different types of breast cancers.
PARP1 functions as a key molecule in the repair of DNA SSBs [17]. PARP1 inhibition leads to persistent SSBs in DNA, which results in the stalling of replication forks and, finally, the formation of DSBs. The effect of this may confer synthetic lethality to cells with defective homology-directed DSB repair, and is therefore emerging as a potential anticancer therapy for cancers with defects in BRCA1/2 or other HR pathway components [14]. Likewise, PARP inhibitors may be useful in tumors that lack mutations in BRCA1 or BRCA2 but have other defects in DNA repair mechanisms [1819202122]. A clear example is the increased sensitivity of XPF/ERCC1-deficient cells to olaparib [23]. It has also been reported that the synergistic activity of platinum compounds in combination with PARP inhibitors could be due not only to HR deficiencies but also to blockade of the repair of platinum intra-strand cross-link lesions by PARP inhibitors [14]. Based on this information and considering that trabectedin blocks NER, we theorized that the combined impairment of two complementary DNA repair pathways (NER inhibition and PARP inhibition) would synergistically increase the sensitivity to the combination of trabectedin and PARP inhibitors [6810]. Additionally, we investigated whether the effectiveness of this approach would be affected by BRCA1 mutations.
Our study suggests that inhibition of NER by trabectedin and of PARP by olaparib has a synergistic effect in causing breast cancer cell death. The highest degree of synergism was observed in MCF7 cells expressing the highest levels of XPG. Conversely, the combination of trabectedin with veliparib or iniparib did not enhance tumor cell death compared with trabectedin or each of these PARP inhibitors alone. A possible explanation for this is that PARP inhibitors differ markedly in their mode of action. For example, recent information reveals that although these drugs inhibit PARP catalytic activity with IC50 values in the low nanomolar range, they exhibit differential cytotoxic activities in three triple-negative breast cancer cell lines, with olaparib being the most active, followed by veliparib and iniparib [2425]. That study did not observe any discernible effect on PARylation for iniparib. This was correlated with modest suppressive effects on cell viability and clonogenic survival [24]. This lack of inhibitory activity was attributed to the fact that iniparib is a prodrug. PARP inhibitors also differ in their ability to trap PARP/DNA complexes. In this sense, olaparib presents a greater potency to produce cytotoxic PARP/DNA complexes compared to veliparib [26]. These results indicate that although PARP inhibitors were assumed to be equivalent based on PARP catalytic inhibition, they are not equivalent with respect to their potency to inhibit PARP.
We also investigated the dynamic effects of trabectedin and olaparib on the cell cycle and DNA damage. The combination of both drugs demonstrated a synergistic effect on DNA damage, as analyzed both by western blotting against phosphorylated γH2AX and a comet assay. When comparing these results, it was clear that the combination induced a higher proportion of DSB than both drugs administered as single agents. Thus, the observed synergistic effect seems to be the result of higher accumulation of DSB after the administration of the combination of trabectedin with olaparib. We analyzed the level of PARylation after single agent or combination treatment, as the covalent modification of proteins by PARylation is a biochemical response to DNA damage [27]. We found that trabectedin induced PARylation in all the breast cancer cell lines, but olaparib inhibited PARylation. The combined treatment of trabectedin and olaparib led to an inhibitory effect, although it was weaker than olaparib alone. These results are interesting, as trabectedin could hyper-activate PARP in breast cancer cells, causing them to become more sensitive to the effects of olaparib. Conversely, PARP inhibition could block the restarting of trabectedin-induced stalled replication forks, thereby possibly causing sustained DNA damage and subsequently potentiating the effect of trabectedin in cells with low DNA repair capacity. Finally, investigation of the cell cycle showed that the combination of both compounds induced stronger levels of G2/M arrest. These increased effects were consistent with the synergistic drug action of the combination.
Interestingly, we did not observe any correlation between BRCA1 status and the response to each treatment alone or the trabectedin/olaparib combination. This could be due to the complex phenotype of the breast cancer cells used in this study or underlying genetic factors other than BRCA1 status (e.g., different expression of XPG, Pol B, or XRCC1). This is not surprising, as Chuang et al. [24] reported that the ability of individual PARP inhibitors to sensitize triple-negative breast cancer cells to cisplatin varied to a great extent in a cell context- and cell line-specific manner. For example, MDA-MB-231 cells that carry a heterozygous mutation in BRCA1 have low endogenous miR-182 and BRCA1 protein expression [28]. Conversely, BRCA1 wild-type MCF7 cells exhibit intermediate levels of miR-182. These levels are similar to triple-negative HCC-38 cells and other luminal-type cells (e.g., T47D or BT483). These cells thus could behave similar to the triple-negative cell lines used in this study. These reasons may all contribute to a lack of hypersensitivity of BRCA1-mutated breast cancer cells to trabectedin, mostly attributed to MCF7 and MDA-MB-231 overexpression of XPG protein.
In summary, we have found that NER blockade with trabectedin and PARP inhibition by olaparib causes selective synergy in breast cancer cell lines, regardless of BRCA1 mutational status. Our findings indicate that this combination induces G2/M arrest and high levels of DNA damage. Most notable is the fact that it is possible to recreate an artificial synthetic lethality by using drugs that affect two different complementary DNA repair mechanisms. Future studies may fulfill the promise of a rationale for artificial synthetic lethality as an approach for the treatment of many common cancers.
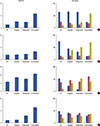
 ), S-phase (
), S-phase ( ), and G/2M (
), and G/2M ( ) cell cycle phases in untreated and drug-treated MCF7 (A), MDA-MB-236 (B), MDA-MB-436 (C), and HCC-1937 (D) breast cancer cells. All cell lines were analyzed after 24 hours exposure to olaparib, trabectedin, and the combination of trabectedin/olaparib as described in Methods section. For (C), only the diploid population is depicted.
) cell cycle phases in untreated and drug-treated MCF7 (A), MDA-MB-236 (B), MDA-MB-436 (C), and HCC-1937 (D) breast cancer cells. All cell lines were analyzed after 24 hours exposure to olaparib, trabectedin, and the combination of trabectedin/olaparib as described in Methods section. For (C), only the diploid population is depicted.




 XML Download
XML Download