

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Breast cancer is the most prevalent type of cancer among women worldwide, representing 25% (1.7 million) of new cases and 15% (more than 0.5 million) of deaths from all cancers [1]. Different therapeutic methods/strategies are commonly employed, including local interventions (surgery/radiotherapy), and systemic treatments (chemo/hormonal therapy or targeted treatments) [2]. However, treated patients can suffer from disease relapse and metastasis [3] due to the presence of a subset of tumor cells known as breast cancer stem cells (BCSCs). BCSCs are a small cell population with unique characteristics such as self-renewal, high proliferation rate, and the ability to generate heterogenic lineages of cancer cells [4], attracting the attention of many researchers for their potential for use as a target for cancer therapy. In this article, the most important characteristics of BCSCs are reviewed.
BREAST CANCER STEM CELLS ORIGIN
Different theories have been proposed about the origin of BCSCs. One of them states that unsuitable regulation or mutations may lead to transformation of dormant normal stem cells to cancer stem cells (CSCs) [5]. According to another, the "misplacement somatic stem cell" theory, CSCs may originate from misplacement of somatic stem cells de novo [6]. Evidence shows that somatic cells can be considered the CSC origin. For example, Mintz et al. [7] indicated the teratogenic effect of somatic cells thorough injection of embryonic somatic cells into a mouse embryo aged 6 days. Several studies suggest that there are intratumoral lineages that have differentiated from common progenitor cells [8].
BREAST CANCER STEM CELL MARKERS
Surface markers, used for the isolation and identification of BCSCs, not only contribute to cell interactions, but also endow them with unique properties. For the first time in 2003, BCSCs were identified and isolated with the CD44+/CD24-/low Lin- phenotype [9]. Since then, the CD44+/CD24- phenotype has been used as a reliable phenotype for the isolation of BCSCs [10111213]. CD44 is a cell surface glycoprotein and specific receptor to hyaluronan. It is a key element for breast cancer adhesion, motion, migration, and invasion [14], and its interaction with osteopontin leads to tumor progression [15]. CD44 has an important role in cell proliferation and tumor angiogenesis [16]. CD24, another surface glycoprotein expressed at low levels, increases a tumor's ability to grow and metastasize [17]. Despite the growing list of CSC markers, some researchers do not consider these markers suitable for identifying CSCs. For example, one report shows that CD44+CD24- is not expressed in all breast cancer cell populations [18].
The other recently recognized marker, aldehyde dehydrogenase (ALDH) [19], consists of a family of cytosolic enzymes involved in the oxidation of intracellular aldehydes and oxidizes retinol to retinoic acid during the differentiation of rudimentary stem cells [20]. ALDH1, the dominant form of the enzyme in mammals, mediates the conversion of retinaldehydes to retinoic acid [21]. The other markers that have been used to identify BCSCs are CD133 [11] and a CD44+ CD49fhi CD133/2hi phenotype found in tumorigenic cells [22]. In vivo and in vitro studies have introduced CD49f [23] and CD61 [24] as BCSC markers as well (Table 1).
SIGNALING PATHWAYS OF BREAST CANCER STEM CELLS
Notch, Hedgehog, and Wnt pathways have been implicated in resistance to therapy and an increased number of BCSCs during/after treatment. These pathways play key roles during embryonic development and adult tissue homeostasis [2526]. Dysregulation of the Notch and Hedgehog pathways, which are involved in normal stem cell self-renewal and differentiation, result in a BCSC phenotype in breast cancer cells [27]. The Wnt pathway plays a pivotal role in stem cell self-renewal and preservation of an undifferentiated state [28].
Hedgehog is an embryonic development organizer pathway that activates Gli1- and Ptch1-positive modulators of the hedgehog pathway, thereby leading to BCSC proliferation [29]. The Notch pathway is important to cell differentiation and connections during both embryogenesis and adulthood. It targets genes that lead to high proliferation and apoptosis inhibition in cancer cells [30]. Examples of transcription factors targeted include cyclinD1, c-myc, CDKN1A, and HES-related repressor protein. This pathway has been reported to act in BCSCs [31]. In addition to signaling pathways, transcriptional factors are significant, too. The main transcriptional factors Sox2, Oct4, and Nanog act as master regulators of pluripotency and maintain the undifferentiated state of cells [32]. Of basal-like breast carcinomas, 43% exhibit Sox2 expression, indicating a less differentiated phenotype [33]. In addition, there is evidence that Sox2 is expressed in derived spheres, those that have been generated from breast cancer tumors and cell lines [34].
Another member of the Sox family, Sox4, induces changes in the epithelial-mesenchymal transition (EMT) process, accompanied by an enhanced number of cells with a CD44+/CD24- phenotype and higher invasion and mobility of cancer cells in vivo and in vitro [35]. In addition, the role of protooncogenes and tumor suppressors is undeniable. They function to coordinately control stem cell self-renewal. For instance, excessive expression of c-myc, KLF4, Oct3/4, and Sox2 results in the differentiation of somatic cells into induced pluripotent stem cells [36]. Nevertheless, proteins encoded by these genes frequently act within less differentiated breast tumors and other tumors [3437]. Evidence suggests that the expression of Oct3/4, Nanog, and Sox2 are strongly associated with different CSCs, including BCSCs [383940].
BREAST CANCER STEM CELLS AND THE EPITHELIAL-MESENCHYMAL TRANSITION
Breast cancer therapy resistance is associated with the phenomenon of EMT and the reverse process, the mesenchymal-epithelial transition (MET) [41]. When BCSCs undergo EMT, the cells that survive chemo/hormonal therapy contain gene determinants similar to those of BCSCs and exhibit epithelial and mesenchymal markers (cytokeratin and vimentin, respectively) [4243]. Following chemo/hormonal therapy, EMT-like gene expression increases, indicating the role of epithelial-mesenchymal plasticity (EMP) in the development of resistance to cytotoxic drugs [424445]. EMP is defined as the ability of cells to undergo EMT and MET. During EMT, a series of changes take place, including the shutdown of transcription and down regulation of epithelial markers such as E-cadherin, and the appearance of mesenchymal markers such as vimentin, fibronectin, and N-cadherin. These changes lead to unstable structures and functions in these cells [46].
In addition, various growth factors, such as epidermal growth factor (EGF), tumor necrosis factor α (TNF-α), and transforming growth factor β (TGF-β), are expressed, which in turn activate mesenchymal transcriptional factors such as ZEB1, SNAIL1, SNAIL2, TWIST1, and TWIST 2. These factors inhibit ZO1, distinct claudins, and E-cadherin [47]. FOXC2, a transcriptional factor that is upregulated in tumors with high stem cell content, is the main marker of mesenchymal stem cells in breast cancer cell lines and of cells undergoing EMT [48]. High expression of SNAIL1, SNAIL2, and TWIST that are expressed during EMT leads to hyposensitivity to paclitaxel, adriamycin, and doxorubicin, respectively, indicating the role of EMP in BCSC therapy resistance [4449]. EMT also results in the resistance of the MCF7 cell line to tamoxifen [50].
MICROENVIRONMENT AND BREAST CANCER STEM CELLS
The specific intratumoral condition location of cells is known as a microenvironment. The microenvironment of CSCs is referred to as a "niche," involving various factors that affect CSC properties. These factors include fibroblast stimuli, immune cells, autocrine signals, and extracellular matrix (ECM) components, as well as physical/chemical factors such as oxygen pressure, nutrients, and PH [51]. Tumor development also depends on cellular communication between different cell populations in a tumor niche [52]. The effects of the microenvironment on CSCs are discussed below.
Microenvironment and growth factors
A vast range of growth factors and cytokines released by tumor cells and cancer associated fibroblasts and immune cells ensure CSC survival and metastasis. Cytokines that are produced include interleukin (IL)-1, -6, and -8, CXCL12, CCL2, and growth factors such as platelet derived growth factor (PDGF), TGF-β, TNF-α, EGF, vascular endothelial growth factor, and FGF [53545556575859606162]. Stimuli that are applied in a microenvironment influence BCSC properties and therapy resistance. For example, IL-6 pathway activity and consequently STAT3/NF-Kβ expression result in trastuzumab resistance and an increased number of HER2 positive BCSCs. IL-6 receptor blockage leads to in vivo inhibition of metastasis and tumor growth [54].
Increased activity of the TGFβ pathway and expression of IL-8, induced by paclitaxel, enhance the cancer stem cell content of triple-negative breast tumors. Blockage of TGF-β type1 and 2 receptors thorough IL-8 inhibition prevents an increase in the number of BCSCs [55]. Moreover, IL-8 receptor (CXCR1) blocking agents, such as certain antibodies or repertaxin, target BCSCs selectively and prevent tumor formation in preclinical models [56].
CXCL12 activates the CXCR4 pathway that is necessary for stem cell survival; therefore, pathway disruption results in less drug resistance. In addition, CXCL12 promotes cancer cell growth through matrix metalloproteinase (MMP)-associated tissue remodeling [575859]. Under the influence of environmental factors (i.e., radiation), the microenvironment produces various types of growth factors, such as IL-1, CXCL12, TNF-α, TGF-β, PDGF, and MMPs. Growth factors facilitate tumor survival and regrowth and endothelial cell survival, and increase the generation rate of invasive CSCs [606162].
Microenvironment and oxygen tension
It is assumed that tissue oxygen pressure can influence CSCs. In oxygenated conditions, cells are active and have high migration and proliferation abilities. In low oxygen conditions (hypoxia) cells appear quiescent and radioresistant. The effect of hypoxia on the biology and physiology of tumors is paradoxical. Hypoxia causes CSC resistance in various ways. First, oxygen is known as a potential radiosensitizing factor. However, hypoxia induces greater radioresistance [63]. Hypoxia is associated with early relapse after radiotherapy, and higher oxygen levels improve patients' responses to treatment [64].
Second, the quiescent state and slower stem cell cycle due to hypoxia leads to greater chemo/radioresistance [65]. Hypoxia, in turn, can activate hypoxia inducible factor (HIF), serving as a CSC survival factor and EMT regulator [6667]. The most prominent HIFs, HIF1, and HIF2, are associated with the Wnt, Hedgehog, and Notch pathways [6869]. HIF1 serves as a key component for BCSC viability, as well as stem cell survival, metastasis, angiogenesis, EMT, and radio/chemo resistance [70]. Enhanced HIF-1α indicates a poor outcome, metastasis, and early relapse of breast cancer patients [71].
THERAPY RESISTANCE MECHANISMS INVOLVING BREAST CANCER STEM CELLS
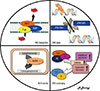
BCSC therapy resistance mechanisms are various, but the most significant of these are described below (Figure 1).
ATP-binding cassette transporters
The ATP-binding cassette (ABC) family of membrane proteins is commonly involved in the transport of compounds and small molecules out of the cell. ABC transporters play an important role in the establishment of chemical homeostasis and cell survival in unfavorable conditions within a broad range of environments, including normal stem cells, the placenta, epithelial cells of the digestive system, and endothelial cells that form the blood-brain barrier [72]. In addition to physiological functions, ABC transporters have been reported to be important in multidrug resistance (MDR) in various cancers [73].
Among the 49 known ABC transporters, three of them have been shown to be MDR regulators, including glycoprotein P (P-gP), multidrug resistance protein 1 (MDR1), and breast cancer resistance protein [74]. These transporters are highly expressed in different cancers, including breast cancer, resulting in cytotoxic drugs being carried out of cells using ATP, and subtoxic drug doses maintained within a cell [75].
Also, overlap among transporters with a vast range of substrates provide tumor drug resistance to a large group of chemotherapy drugs, including antimetabolites, topoisomerase inhibitors, and taxanes, as well as tyrosine kinase inhibitor drugs, such as erlotinib, imatinib, gefitinib, nilotinib, and sorafenib, that serve as molecular targeting drugs [7476]. Studies have shown that preventing ATP-binding cassette G member 2 (ABCG2) transporter activity using Ko143 (fumitremorgin-type indolyl diketopiperazine) and phosphodiesterase-5 inhibitors can improve the performance of anticancer drugs [7778].
Aldehyde dehydrogenase activity
ALDH is an enzyme family involved in the oxidation of intracellular aldehydes to carboxylic acids, retinoic acid, and γ-amino butyric acid (GABA) biosynthesis, with a significant role in the survival and differentiation of normal and CSCs [7980]. ALDH induces CSC radioresistance both through direct removal of oxygen radicals that are produced and indirect production of an antioxidant compound, nicotinamide adenine dinucleotide (phosphate) [81].
In addition, ALDH1 seems to be associated with breast cancer malignancy, metastasis, and invasion [82]. High activity of ALDH1 and 3 isoforms enables the cells to metabolize cyclophosphamide and analogues such as ifosfamide, mafosfamide, and 4-hydroperoxy cyclophosphamide, and to detoxify aldophosphamide (an intermediated product) to carboxyphosphamide [8384]. ALDH+ cells not only exhibit paclitaxel and epirubicin resistance but also increase in number after chemotherapy [85].
DNA repair
Radiotherapy and various types of chemotherapy drugs damage DNA through various mechanisms, such as DNA synthesis inhibition (methotrexate), topoisomerase inhibitory action (daunorubicin, doxorubicin), and DNA cross-link formation (carboplatin, cisplatin, oxaliplatin), in which DNA repair failure results in cell death [86]. CSCs possess DNA repair and reactive oxygen species (ROS) scavenging mechanisms [25]. Nevertheless, DNA damage and repair mechanisms are generally different. In addition, DNA damage as an inducing factor may activate checkpoint mechanisms. DNA repair is proposed to be acting in various types of tumors, including human and mouse breast tumors [8788]. The most lethal type of DNA damage is a double-strand break, which is repaired in two ways [89].
Homology-directed recombination (HDR) is an error free repair mechanism. The recombination steps (catalyzed by distinct enzymes) include 3'-5' resegmentation of the two ends of DNA, formation of single-strand DNA at the 3' end, assembly of RAD51 filaments (a protein family contributing in the repair of DNA double-stand breaks), and finally repair synthesis by annealing at the end of the double-strand break [9091]. Since this method requires intact sister chromatids, HDR repair occurs only during the S and G2 phases of the cell cycle.
Unlike HDR repair, nonhomologous end joining (NHEJ) introduces error. In this process, the KU70/KU80 proteins join the ends of DNA strands. In addition, nucleases, polymerases, DNA-dependent protein kinases, and ligases participate in the NHEJ repair process [91].
Regarding DNA damage-activated checkpoint mechanisms, two pathways are noticeable. ATR-Chk1 and ATM-Chk2 are kinase-signaling pathways activated by double-strand and single-strand DNA breaks, respectively. DNA damage checkpoint signaling makes DNA repair possible thorough cell cycle inhibition [92]. The ATM/ChK2 activating pathway has been proposed to be associated with BCSCs radioresistance, and the existence of the ATM inhibitor and radiosensitivity enhancer, KU55933, suggests that the ATM pathway is an appropriate target for eliminating radioresistance in BCSCs [93].
Reactive oxygen species scavenging
Different levels of oxygen are essential to promoting intracellular reactions. ROS, active radicals that are produced during oxygen metabolism, participate in the regulation of physiologic events of the cell, such as proliferation, migration, wound healing, and angiogenesis [94]. Excess amounts of ROS produced in response to radiation, followed by interactions with cell components like DNA, proteins, and lipids, induce cell death [95].
Both normal and cancer cells establish an equilibrium between production and depletion of ROS using compounds such as catalase, glutathione peroxidase, superoxide dismutase, and thioredoxin [96]. BCSCs have specific mechanisms to guard against the genotoxic effects of ROS, including more effective ROS scavenging and lower levels of ROS production. In addition, genes encoding the proteins superoxide dismutase, catalase, and glutathione peroxidase enzymes, all of which are involved in ROS scavenging, are upregulated in BCSCs significantly. The importance of ROS scavenging appears when tumor treatment with buthionine sulfoximine (BSO) leads to lower radioresistance with decreased clonogenic potential of CSCs. BSO seems to promote this process by inhibiting glutamate cystein ligase [97].
METASTATIC ROLE OF BREAST CANCER STEM CELLS
Metastasis is a complicated process that begins with the migration of cancer cells from the primary tumor and is followed by regional invasion and entry to the circulatory system. Eventually, metastasis terminates with the arrest of cells at a secondary site, extravasion, and colonization. Colonization is not merely a single term; it involves a series of events, including survival of cancer cells until they enter the tissue, the formation of micrometastases, a latency phase, regrowth from latent micrometastases, progressive tumor growth surpassing host tissue growth, and recirculation and formation of tertiary lesions in the same or different organs [98].
The most common site for breast cancer metastasis is bone, but other organs such as the lung and liver are also involved [99]. Hyaluronan and osteopontin, the major components of breast cancer target tissues (bone, brain, liver, and lung), serve as specific ligands for CD44 [100]. Osteopontin, which is expressed in different tissues, contributes to cell adhesion and immune responses [101102]. Breast cancer metastatic cells attach to the bone marrow endothelium via osteopontin [103]. Osteopontin is also associated with a higher incidence of tumor metastasis and invasion [104]. Highly expressed osteopontin, IL-1, and CXCR4 enhance the metastatic potential of breast cancer cells [105].
In addition to osteopontin, one of the protein components of ECM, tenascin C, enhances the efficiency of signaling pathways Wnt and Notch, stabilizing breast cancer-initiating tumor cells in the lungs [106]. Breast cancer cells exhibit high Src tyrosine kinase activity, leading to P13K-Akt pathway sensitivity associated with cell survival. The sensitized P13K-Akt pathway, in turn, is activated by CXCL12 and IGF1, which are released from the bone marrow stroma [107].
Vascular cell adhesion molecule 1 (V-CAM1) is highly expressed in breast cancer cells, sensitizing the P13K-Akt pathway to external signals. Moreover, α4β1 integrins belonging to tumor-associated macrophages activate ezrin (adaptor protein for P13K and Akt) via V-CAM1 [108]. MMP1, which facilitates metastasis to the brain, has a high expression in cells that metastasize to brain. MMP1 breaks down occludin and claudin, the main components of the blood-brain barrier. Also, cyclooxygenase2 (COX2) upregulation induces prostaglandins that promote MMP1 expression directly. Astrocytes that have been activated by COX2 and prostaglandins produce chemokine ligand 7 (CCL7), which is involved in the progression of tumor-initiating cells in the brain [109].
AUTOPHAGY AND ITS ROLE IN CANCER AND BREAST CANCER STEM CELLS
CSCs may be subjected to unfavorable conditions, such as hypoxia, loss of nutrients, or toxic drugs, in their microenvironments. To resist these conditions, CSCs possess various catabolic processes to maintain their viability and metabolic homeostasis. The main mechanism for homeostasis maintenance is autophagy [110]. During autophagy, proteins or organelles form autophagosome-joining lysosomes that become autophagolysosomes. After this, lysosomal enzymes such as cathepsins break down their contents to return the energy or amino acids to the cells [111].
Autophagy failure is associated with muscle atrophy, degeneration of the nervous system, and a broad range of cancers [112]. In investigations of the role of autophagy in cancer, a dual function has been observed. On the one hand, it prevents the accumulation of damaged proteins and organelles, serving as a tumor suppressor, and on the other hand, it acts as a tumor enhancer with hypoxia or the loss of nutrients. Cell stress or increased metabolic requirements force tumor cells to activate autophagy [113].
Radiotherapy suppresses breast, prostate, glioblastoma, and colon cancers by enhancing autophagy hyperactivity of cancer cells and autophagy-induced cell death [114115]. In addition, D-cyclovirobuxine induces autophagy-dependent death in the MCF7 cell line [116]. Nevertheless, the main role of autophagy is maintaining of tumor cells in stressful conditions. Autophagy is a mechanism for preserving cell integrity under cytotoxicity, metabolic stress, or radiation-induced disruptions [114117118119].
Accordingly, loss of autophagy results in increased DNA damage and chromosomal disruption in cancer cells during stress [117118]. Autophagy inhibition causes higher radiosensitivity of breast, prostate, glioblastoma, and colon cancers [114119]. Studies have shown that autophagy markers LC3B, Atg5, and Atg12 are involved in autophagosome formation, and are expressed at high levels in breast cancer stem-like quiescent cells [120].
Autophagy inhibition prevents the invasion of breast cancer progenitor cells, as well as spheroid and xenograft tumor formation. Autophagy activity seems to be significantly higher in mammospheres than in a control group [121]. Moreover, autophagy has been reported to be higher in an ALDH+ population of no special type breast cancer [122]. Chloroquine can inhibit autophagy through the prevention of lysosomal mass breakdown. It also interferes with E-cadherin endocytosis, thereby inhibiting EMT [123].
DNA METHYLATION AND BREAST CANCER STEM CELLS
DNA methylation is a process in which methyl groups are added to the 5' end of cytosine residues in the guanosine cap. In mammals, DNA methylation is accomplished by three types of DNA methyl transferase enzymes (DNMT1, 3a, and 3b). DNMT3a and DNMT3b serve to regulate the methylation patterns of genes [124]; therefore, they play a key role in the regulation of stem cell properties by de novo methylation [125]. DNMT3b seems to play an even more significant role in this respect, inducing aberrant DNA methylation patterns and maintaining the undifferentiated state of CSCs [126]. Exploring 68 methylated regions of BCSCs, hypomethylation pattern was reported to be more common in these regions than in nonstem cells, and to be associated with poor prognosis [127].
MICRO RNAs AND BREAST CANCER STEM CELLS
Micro RNAs (miRNAs) are noncoding regulatory RNAs contributing to posttranscriptional regulation of gene expression. miRNAs result in mRNA degradation through the inhibition of ribosome activity, mRNA translation, and deletion of the 3' end of the poly A tail and the 5' end of the guanosine cap [128]. miRNAs restore stem cell characteristics to those of normal and cancer stem cells; hence, their dysregulation is associated with tumorigenicity.
Several examples of the involvement of miRNA in tumorigenicity have been reported. miR21 is an oncogene miRNA that is involved in breast cancer EMT and maintains stem cell-like characteristics [129]. miR200c suppression appears to act in BCSC tumorigenicity [130]. miR9 is associated with a BCSC phenotype and EMT state [131]. miR203 loss is observed in the majority of BCSCs. Finally, miR203 re-expression induces differentiation and suppression of stem and mesenchymal cell properties [132].
CONCLUSION
In recent years, the cancer stem cell theory has led to new insights into cancer treatment. Among different types of cancers, breast cancer has received the most attention due to the number of people impacted. The isolation of BCSCs from a solid tumor, along with an extensive understanding of cellular, molecular, and signaling pathway mechanisms, enforce the belief that therapy failure and therapy resistance are due to the presence of cancer stem cells, a subpopulation that has high proliferative and metastatic potential and that can act as a reservoir for tumorigenicity.
BCSC function is remarkable in several aspects. First, this population is resistant to conventional therapies due to enhanced membrane transport by specific protein transporters, specific mechanisms of DNA repair, and ROS scavenging systems, and the ability to detoxify cytotoxic drugs. Second, transcriptional factors, signaling pathways, and tumor suppressor genes act to maintain and amplify a state of stemness. Third, extensive interactions among cancer stem cells, their microenvironments, and other cells present initiate a cascade of growth factors and inducing elements, which in turn influence cancer stem cell function. More studies are needed to investigate each of these aspects of BCSCs.

 XML Download
XML Download