

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Despite earlier diagnosis and advances in treatment, breast cancer is a heterogeneous disease that remains a leading health threat to women [1]. Recently, several studies revealed granulocyte colony-stimulating factor receptor (G-CSFR) expression in tumor cells or autocrine secretion of granulocyte colony-stimulating factor (G-CSF) expression in nonhematopoietic tumors such as colon cancer, ovarian cancer, squamous cell cancer, malignant melanoma, and sarcoma [234567891011]. These studies highlight the importance of developing more robust methods to treat and prevent breast cancer. G-CSF has been shown to stimulate proliferation and angiogenesis, and it subsequently enhances malignant potential [210].
However, functions of G-CSF in normal epithelia are different from its roles in cancer. G-CSF is a 25-kDa glycoprotein synthesized by a variety of cell types that could stimulate proliferation of bone marrow granulocytic progenitor cells and promotes their differentiation into granulocytes [12]. In addition, G-CSF can exert effects on cells outside the granulocyte lineage. For example, G-CSF can cause proliferation in vitro or monocytic differentiation of some myeloid leukemias as well as growth and migration of endothelial cells [1314]. It was reported that these diverse biological effects were mediated by the interaction of G-CSF with a cell-surface receptor expressed on responsive cells [15].
Recombinant human G-CSF (rhG-CSF) is used to treat patients with a variety of neutropenias and could function by binding to its cognate cell surface receptor, which is a member of the cytokine receptor superfamily [1617]. Studies have shown that stimulation of granulocytic cells with G-CSF leads to the activation of the Janus kinase/signal transducers and activators of transcription (JAK/STAT) and Ras/mitogen activated protein kinase (MAPK) pathways [1617181920]. How G-CSF promotes breast cancer is unknown; however, increased G-CSF signaling is linked to trastuzumab resistance in erbB2-positive breast cancer [21]. In this study, we assessed the ability of rhG-CSF to independently initiate mammary tumorigenesis and/or accelerate erbB2-mediated mammary tumor growth progression. Because of the potential risk of stimulation of proliferation by rhG-CSF, information concerning G-CSFR expression in tumor cells would be helpful for treating cancer patients.
To explore how G-CSF contributes to breast cancer development in vivo, we generated multiple lines of human breast cancer cells that contain human macrophage chemokine factor 7 (MCF-7), SKBR-3 and MCF-10A, that expressed G-CSFR, which has been reported in other solid cancers. Its expression was induced in mouse mammary gland tumors, which highlighted the similarities between human disease and mouse models. We investigated the cellular and molecular mechanisms of G-CSF action in breast cells using immunohistochemistry (IHC) and real-time polymerase chain reaction (RT-PCR). We also examined the regulation of a number of G-CSF genes, including critical growth and cell cycle-regulating genes using breast cell lines and mammary gland samples from mice treated with rhG-CSF. Our study may provide a novel foundation for future breast cancer treatment.
METHODS
Cell lines and culture conditions
Breast cancer lines MCF-10A, MCF-7 (HER2 negative), and SKBR-3 (HER2 strongly positive) were obtained from Jenniobio Biotechnology Co., Ltd. (Guang Zhou, China). MCF-10A is normal breast epithelial cell line. Her-2 is a proto-oncogene epidermal growth factor receptor and an important prognostic factor in breast cancer. SKRB-3 is an erbB2-high expression cell line, which is consistent with mouse mammary tumor virus-erbB2 (MMTV-erbB2) models. MCF-10A and SKBR-3 cells were cultured in Roswell Park Memorial Institute (RPMI)-1640 medium (Hyclone, Logan, USA); MCF-7 cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) (Hyclone) supplemented with 10% fetal bovine serum (FBS) at 37℃ in a 5% CO2 incubator according to the recommended protocols. Western blot was performed to detect protein expression.
Cell viability assay
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay was used to assess cell viability. Cells were seeded onto 96-well plates at a concentration of 2×103 per well, incubated overnight under standard culture conditions, and exposed to rhG-CSF (Amoytop, Xiamen, China) at various concentrations. MTT solution (20 µL) was added to each well after 24, 48, and 72 hours. The plates were incubated for 2 hours in an incubator and oscillated for 10 minutes. The absorbance was measured at 540 nm, with 655 nm as the reference wavelength. All experiments were performed in triplicate.
Transgenic mice
This study was approved by Henan International Science and Technology Cooperation Program. The study followed the recommendations of the National Institutional Animal Care and Use Committee for animal handling, maintenance, treatment, and sacrifice. The MMTV-erbB2 transgenic mice used in this study were purchased from Jackson Laboratory (Bar Harbor, USA; license number: 0207 AX1). The mice were kept in a specific pathogen-free room (n=3 per cage), maintained at an ambient temperature of 22℃±2℃ and humidity of 50%±10%, with a 12-hour light/12-hour dark cycle. Mice had ad libitum access to tap water and food.
Sixty 12 weeks old healthy adult female MMTV-erbB2 transgenic mice that weighed approximately 13 g (11-15 g) were selected for this study. The transgenic mice were randomly divided into three groups: low-rhG-CSF group (rhG-CSF, 0.125 µg), vehicle-rhG-CSF group (normal saline, 0.25 µg), and high-rhG-CSF group (rhG-CSF, 0.25 µg). These groups are similar to those used in human trials; the peak serum concentrations of G-CSF after administration of a standard dose of G-CSF (5 µg/kg) was found to range from 15 to 30 ng/mL [2223]. Four inguinal mammary glands were obtained, spread onto slides, and fixed with 4% formaldehyde overnight after 2 months of treatment. The glands were placed in 70% ethanol for 1 hour followed by water for 30 minutes. The slides were then stained with 0.2% carmine overnight. The mammary glands were dehydrated sequentially in 70%, 90%, and 100% ethanol for 30 minutes each and then cleared in toluene to dissolve fat in the gland. The slides were maintained in methyl salicylate for analysis under light microscope (Nikon, Tokyo, Japan).
Mice were observed once every 3 days from 20 weeks of age to monitor tumor development. Tumor incidence and latency period were calculated based on the date when a tumor was detected by palpation of the mammary gland (tumors were detected when the diameter was >3 mm). The largest maximum diameter was recorded once every 3 days from the day when single or multiple tumors were first detected. Mice were euthanized using pentobarbital sodium when the volume of the breast tumor was 1.5 cm2 or the mouse was 60 weeks of age. The mammary glands of specimens were divided into two parts: one part was immediately kept in liquid nitrogen and then transferred to a -80℃ refrigerator, and the other part was fixed in 10% neutral formalin (pH=7.2) for 24-48 hours at room temperature. Mammary epithelial pellets from four mice were pooled into one sample and were used for RNA isolation. We also examined a number of G-CSF regulation genes, including critical growth and cell cycle-regulating genes using breast cell lines and mammary gland samples from rhG-CSF-treated mice.
Immunohistochemical analysis
Proliferating cell nuclear antigen (PCNA), cluster of differentiation (CD34), and signal transducer and activator of transcription (STAT3) expression was evaluated by IHC staining. Tissue slides were dewaxed in xylene and then rehydrated through graded ethanol. Tissue slides were incubated overnight with PCNA (1:500; Boster, Wuhan, China), CD34 (1:500; Boster), and STAT3 (1:200; Cell Signaling Technology, Shanghai, China) antibodies at 4℃. Antibody binding sites were visualized by using the Ultra Sensitive™ S-P Immunohistochemistry kit (Beijing Zhongshan Golden Bridge Biotechnology, Beijing, China). Finally, sections were counterstained with 0.1% hematoxylin. The percentage of cancer cells with positive staining was evaluated. IHC staining was evaluated by two independent pathologists who were blinded to the clinical data. In this study, the scoring patterns for PCNA, CD34, and STAT3 staining were as follows: 0, negative staining for all cells; 1+, weakly positive for cytosolic staining in <10% of cells; 2+, moderate-to-strong positive staining covering between 10% to 50% of cells; and 3+, strongly positive staining for >50% of cells. IHC scores were grouped into two groups, low expression (0 and 1+) and high expression/over expression (2+ and 3+).
RNA extraction and quantitative RT-PCR
Total RNA was extracted using Trizol reagent (Invitrogen Life Technologies, Carlsbad, USA) according to the manufacturer's protocol. The primer sequences for quantitative RT-PCR (qRT-PCR) amplification are shown in Supplementary Table 1 (available online). qRT-PCR was performed using the Fast Start Universal SYBR Green Master (ROX; Roche, Toronto, Canada) on the Bio-Rad CFX96 qRT-PCR detection system (Applied Biosystems Inc., Foster City, USA). The CFX Manager software was used to calculate a threshold cycle (Ct) value for β-actin and G-CSF during the log phase of each cycle. Expression data were normalized to the geometric mean of the housekeeping gene β-actin to control expression variability and then analyzed using the 2-ΔΔCt comparative Ct method, where ΔΔCt=ΔCt G-CSF-ΔCt β-actin. Each sample was tested in triplicate to minimize experimental variability, and the mean femtogram expression level was calculated.
Statistical analysis
Statistical analysis was performed with SPSS version 19.0 statistical software package (IBM Corp., Armonk, USA). We used Bonferroni's multiple comparison test (MCT) to analyze normally distributed data, and the Kruskal-Wallis test followed by Dunn's MCT to analyze data that were not normally distributed. Multiplicity was summarized as mean and SE and then compared by one-way analysis of variance. The number of mammary glands with preinvasive and invasive lesions were counted and analyzed by Fisher exact test. The Kruskal-Wallis rank sum test was used to compare the control, low-dose, and high dose groups. p<0.05 was considered statistically significant; p<0.01 was considered highly statistically significant.
RESULTS
rhG-CSF promotes malignant mammary epithelial cell proliferation
MCF-10A, MCF-7, and SKBR-3 cells were treated with rhG-CSF (0, 0.01, 0.1, 1, 10, 100, and 1,000 µg/L), and their growth rates were measured (Figure 1A). G-CSFR expression was measured by Western blot (Figure 1B). The results showed that rhG-CSF promoted cell growth, and it had a more profound effect in MCF-7 and SKBR-3 cells than in MCF-10A cells (p=0.002 and p=0.001, respectively). In particular, in MCF-7 cells, when the concentration was 0.1 µg/L, cell proliferation significantly increased, but then decreased with increasing concentration. For SKBR-3 cells, when the concentration was 1 µg/L, the cell proliferation promotion effect was significant. The results showed that rhG-CSF had little effect on normal breast epithelial cells, but it promoted the proliferation of MCF-7 and SKBR-3 cells.
rhG-CSF promotes mammary carcinoma development
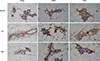
Our results showed that, compared with the control group, both rhG-CSF 0.125 µg and rhG-CSF 0.25 µg promoted pubertal mammary gland development. Whole mounts showed that chronic exposure to low doses of rhG-CSF significantly promoted pubertal mammary gland development (Figure 2). Further, all of these effects were absent in the higher, regulatory-based doses of rhG-CSF. No significant differences between treatments were noted for location, incidence of necrosis, or grade of the developing tumor.
An estrogen receptor (ER)-positive mammary tumorigenesis model that simulates oncogenic events in human breast cancer was chosen for this study to determine the cancer-promoting effect of rhG-CSF [24]. MMTV-erbB2 mice carry the inactivated, wild-type erbB2 proto-oncogene, whose expression is targeted to breast tissue by transcriptional control of the MMTV promoter.
Figure 3 shows that low-dose treatments with rhG-CSF did not significantly promote tumor formation. No cutaneous toxicities, weight loss, appetite loss, or skin erythema were observed in mice treated with either rhG-CSF dose.
rhG-CSF promotes mammary epithelial cell proliferation and increases PCNA and STAT3 expression
To determine the mechanism of rhG-CSF-induced growth, we examined the effect of rhG-CSF on proliferation by measuring PCNA and STAT3 expression in mammary glands obtained after 4 months of treatment. rhG-CSF significantly promoted epithelial cell proliferation by 80% (p=0.001), as determined by PCNA IHC staining (Figure 4). Figure 4 shows the IHC results of PCNA, STAT3, and CD34 from consecutive sections. The percentage of PCNA-positive cells showed substantial variation in the vehicle group ranging from 80% to 5%; however, the median amount was 36.8%. In some samples, the high PCNA levels may be the result of hyperplasia development in erbB2-expressing mammary cells. No statistical difference was observed in STAT3 levels between vehicle and rhG-CSF treatments (data not shown), which indicates that rhG-CSF promoted mammary tumorigenesis, primarily by promoting proliferation. The positive Spearman correlation coefficients were statistically significant in both vehicle and rhG-CSF groups.
Chronic rhG-CSF exposure alters RNA expression of signaling pathways involved in carcinogenesis
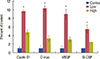
The 112-day-old MMTV-erbB2 mice that were exposed to low rhG-CSF doses exhibited a dose-dependent increase in cell proliferation. rhG-CSF exposure also caused a dose-dependent increase in the promotion index in mammary epithelial cells (Figure 5). The results showed that the low-dose group (rhG-CSF 0.125 µg) had a significantly greater promotion index compared with the vehicle group. To ensure that rhG-CSF did not promote tumorigenesis by driving transgene expression, we investigated erbB2 expression (Figure 5). No differences were observed in RNA expression of erbB2 between any of the treatment groups. G-CSF expression in the low-dose group was significantly increased compared with that of the control group (p=0.006). Vascular endothelial growth factor (VEGF) expression was not altered by any of the treatments. We measured the expression of the erbB2 ligand that is reported to be under the control of cyclin D1, myelocytomatosis (C-myc), and found a dose-dependent increase in expression. These results indicate that, in this mouse model, rhG-CSF promotes mammary tumorigenesis through proliferation in normal and premalignant tissues.
DISCUSSION
Several growth factors play pivotal roles in cell proliferation, migration, and differentiation [25]. With the advent of chemotherapy and radiotherapy, rhG-CSF has played an important role in the treatment of neutropenia in cancer patients since the 1990s [26]. Our results, combined with those from other studies, support the idea that the tumors in MMTV-erbB2 transgenic mice can arise through a multistage process promoted by rhG-CSF. The data we obtained are consistent with previous reports, which demonstrated that rhG-CSF promotes premalignant lesion development and decreases PCNA and STAT3 expression [27]. Moreover, it was found that rhG-CSF promoted mammary epithelial cell proliferation [21].
Moon et al. [28] were the first to reveal proliferation and differentiation in leukemia and solid tumor cells using 38 solid tumor cell lines and five hematopoietic cell lines. They found that G-CSF stimulated proliferation (40%-80% increase in proliferation in treated cells compared with that in control cells) in three leukemic cell lines and induced differentiation of AML1/ETO+ leukemic cells. Among the 38 solid tumor cell lines, five cell lines (hematopoietic; 2 breast carcinoma, HCC 1954 and MCF-10A; squamous cell carcinoma of the larynx; and melanoma cell lines) showed G-CSFR mRNA expression [28]. It is worth noting that our findings did not contradict previous reports, but rather provided more extensive data.
rhG-CSF promoted malignant mammary epithelial cell proliferation; in particular, when the concentration of was 0.1 µg/L, there was a significant increase in cell proliferation. Moreover, low doses of rhG-CSF also significantly promoted pubertal mammary gland development compared with the control group. Previous studies of breast cancer reported that low-dose G-CSF is efficient for maintaining neutrophils and white blood cell counts at safe levels. Exogenous G-CSF could activate the G-CSF signaling pathway to significantly promote cancer cell proliferation, enhance STAT3 phosphorylation, and promote expression of a downstream gene to increase the number of cancer cells [29]. G-CSF was reported to be closely associated with human cancer. In ovarian cancer, G-CSFR was predominantly expressed in high-grade serous ovarian epithelial tumor samples and a subset of ovarian cancer cell lines [27]. Stimulation of G-CSFR-expressing ovarian epithelial cancer cells with G-CSF led to increased migration and survival, including resistance to against chemotherapy-induced apoptosis, and the effects of G-CSF were mediated by signaling via the downstream JAK2/STAT3 pathway [27].
Our study revealed that the first chronic rhG-CSF exposure changed RNA expression in the signaling pathways. The role of G-CSF in promoting breast cancer is very complex, and elucidating its specific functions in the signaling pathway requires further research. Studying G-CSF will provide a better foundation for improving clinical treatment of different types of breast cancer.
Solid tumors of nonhematopoietic origin have been shown to express G-CSF or G-CSFR [30]. Cell differentiation and proliferation were stimulated by the interaction between G-CSFR expression of malignant cells and G-CSF. The condition of the patient can be monitored for both cell proliferation and differentiation by detecting G-CSFR expression in primary tumor cells. Many studies have confirmed G-CSFR expression in a variety of solid tumor cells in lung cancer, colon cancer, ovarian cancer, head and neck squamous cell carcinoma, malignant melanoma, and sarcoma [234567891011]. How G-CSF is involved in solid tumor formation and progression is not fully understood. Our findings indicate that G-CSF is a critical factor in enhancing the breast tumor formation. rhG-CSF has a potential risk for use in breast cancer therapy, particularly aggressive types of breast cancer, because it interferes with the process of metastasis; however, this requires a further study.
Our findings revealed that G-CSFR expression might be an effective therapeutic target to counter breast cancer metastasis. Additionally, it could be used either alone or in combination with other effective therapeutic approaches. These results provide strong preclinical and experimental evidence that G-CSF is a critical factor that promotes breast tumorigenesis, and it is a potential therapeutic target for aggressive breast cancer.



 XML Download
XML Download