

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Breast cancer, as the most common malignancy, is the major cause of cancer-related deaths of women worldwide [1-3]. Age plays a critical role in the incidence of this type of cancer, as it has been found that young breast cancer patients have worse outcomes than older premenopausal or postmenopausal patients. A large number of studies point out the fact that the survival rates in patients aged up to 34 years is very poor. Women falling in the age group of 40-49 have the best prognosis [4,5]. Data suggest that hormonal mechanisms may play key roles in this age prognosis relationship, as the difference in survival patterns between age groups were seen only in patients with hormone receptor positive tumors and not in those with hormone receptor negative tumors [5]. Epidemiological data have shown that the incidence of breast cancer in women is closely related to a high fatty diet. Furthermore, breast cancer cells have significant lipogenic capacity, and inhibition of fat metabolism in these cells is associated with their growth arrest and apoptosis [2,6].
Breast cancer treatment involves surgery, chemotherapy, radiation therapy, hormonal therapy, or combination therapy [1,7]. Breast cancer tumors, which are estrogen/progesterone receptor (ER/PR)-positive, are 60% more likely to respond to hormonal therapy, whereas ER/PR-negative tumors show only 5% to 10% response to hormonal therapy [7]. Hormonal therapies for breast cancer are usually done after surgery, chemotherapy, and/or radiotherapy. This therapy is designed to help prevent the recurrence of the disease by blocking the effects of estrogen. Tamoxifen, a drug taken by some women for up to 5 years after the initial treatment of breast cancer, helps in preventing the recurrence of tumor by blocking the ERs on breast cancer cells. The role of hormonal therapy drugs is to slow down the growth rate of cancer cells, which are growing in response to the presence of estrogen and its receptor [7,8].
Radiotherapy is an important treatment modality in the management of cancer. Nearly 60% of all cancer patients are treated with radiation [9], either alone or in combination with surgery and chemotherapy [10-12].
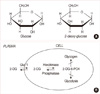
Unfortunately, the efficacy of conventional radiotherapy is limited by 1) the presence of hypoxic, intrinsically radio-resistant, and repair-proficient tumor cells; 2) genetic, metabolic, and microenvironmental heterogeneity of tumors [10]; and 3) undesirable damage to the normal healthy tissues [10,11,13,14]. Therefore, significant improvement in therapeutic efficacy can be achieved only by developing effective approaches based on a comprehensive understanding of the radiobiology of the tumor and normal tissues, to selectively enhance the radiation damage in tumors while reducing the damage to normal tissues [10,11,15]. Radiotherapy is now of routine value after conservative surgery to reduce locoregional tumor recurrence. However, some cancer cells are intrinsically resistant to ionizing radiation induced damages, and such a treatment can actually induce tumor cell proliferation and repopulation, resulting in a diminished response to radiation and poor tumor local control. More important, hypoxia has been associated with drug resistance and reduced sensitivity to radiation therapy, partly because of the upregulation of hypoxia inducible factor 1 (HIF-1) and activation of survival molecules such as Akt and nuclear factor kappa-light-chain-enhancer of activated B cells (nuclear factor-κB). Therapeutic resistance associated with hypoxia is a significant problem in the clinical treatment of cancer, and inhibition of glycolysis may provide a novel approach to overcoming such a resistance. In fact, some studies showed that, under hypoxic conditions, cells exhibited increased sensitivity to the glycolytic inhibitor, 2-deoxy-D-glucose (2-DG) [16]. Hexokinase, the first enzyme in the glycolytic reaction, may be the key regulator in 2-DG induced apoptosis [14,16,17], as it causes glucose to undergo metabolism (Figure 1). Recently published data support the role of hexokinase activity in the prevention of apoptosis mediated by Akt [14,17]. It is also unregulated by HIF and, therefore, the cell may become more resistant as there are more enzymes to be inhibited by a given amount of 2-DG [18].
Accelerated glucose uptake [19] during anaerobic glycolysis (Warburg effect) [14-17,20-26], and loss of regulation between glycolytic metabolism and respiration [27], are the major metabolic changes found in malignant cells. In general, cancer cells have increased rates of glycolysis as well as pentose-phosphate cycle activity and slightly reduced rates of respiration [24,25,27]. Enhanced glucose uptake and glycolysis in tumors arise as a result of multiple reasons including oncogenic transformation-linked alterations in gene expression, mitochondrial mutations, and hypoxia in the case of solid tumors, which result in enhanced levels and activities of glucose transporters and glycolytic enzymes [14,16]. Studies have shown that glucose deprivation can induce cytotoxicity in transformed human cell types via metabolic oxidative stress. In addition, transformed human cell types appear to be more sensitive to glucose deprivation induced cytotoxicity and metabolic oxidative stress than non-transformed human cell types [20]. Glucose analogues have been found to profoundly inhibit glucose metabolism in cancer cells in vitro and in vivo [14,17,20,21]. Of the many glucose analogues that have been investigated, 2-DG has been proven to be the most effective in inhibition of cell metabolism and adenosine triphosphate (ATP) production [14,17,20,21]. 2-DG is a structural analogue of glucose differing at the second carbon atom by the substitution of hydrogen for a hydroxyl group (Figure 2A) and appears to selectively accumulate in cancer cells by metabolic trapping because of increased uptake, high intracellular levels of hexokinase or phosphorylating activity, and low intracellular levels of phosphatase (Figure 2B) [17,21]. 2-DG undergoes facilitated diffusion into cells via glucose transporters. Once inside the cell, 2-DG is phosphorylated by hexokinase to 2-deoxy-D-glucose-6-phosphate (2-DG-6-P). 2-DG-6-P is not a substrate for glucose-6-phosphate dehydrogenase or phosphohexoisomerase. Therefore, once formed, 2-DG-6-P is not further metabolized, and, therefore, the output from glycolysis and the pentose phosphate pathway gets reduced, and 2-DG-6-P will accumulate in the cell until dephosphorylated by phosphorylase [16-18,23].
2-DEOXY-D-GLUCOSE FUNCTION
Cellular processes leading to the error-free repair and fixation of DNA lesions require a continuous flow of metabolic energy, which is frequently supplied by enhanced glycolysis in cancer cells. However, in normal cells, the respiratory pathway is the major contributor to energy (ATP) production [10,15,16,28]. Two properties of 2-DG, namely, the inhibition of glycolysis and the preferential accumulation in cancer cells, have formed the basis for further investigating the mechanism of 2-DG for its use as an antitumor agent. It has been speculated that, cancer cells, which are initially treated with 2-DG, exhibit a stress response caused by a depletion of intracellular energy [16,17,27]. The stress response results in increased levels of glucose transporter expression and increased glucose uptake, which allow more 2-DG to enter the cell. As a consequence of high intracellular 2-DG concentrations, hexokinase and hexose phosphate isomerase are inhibited; energy stores such as ATP are further depleted; and finally, the cell activates the cell death pathway [16,17]. In addition, increased pro-oxidant production and profound disruptions in thiol metabolism consistent with metabolic oxidative stress were also noted in cancer cells during glucose deprivation or when treated with the glucose analogue 2-DG [27].
However, malignant transformation of cultured cells with oncogenes or oncoviruses results in an absolute increase in the amount of glucose transported into the cell. This is mediated by transcriptional activation of the GLUT1 glucose transporter gene resulting in increased levels of glucose transporter mRNA and protein. GLUT1 protein expression is increased in cancer cells and has been reported to increase during cellular stress and also during glucose deprivation [17,18].
Studies have shown that the cytotoxic effect of 2-DG is heterogeneous among different tumor cell lines. While profound growth inhibition and cell death have been found in some cells, a marginal effect on growth and clonogenicity have also been reported in a few. A number of factors contribute to these two diversified responses, which includes the extent of glucose dependence and glycolysis, energy deprivation in the form of ATP depletion and imbalance in the oxidative stress (mitochondrial metabolism) [23], levels of glucose transporters, c-Myc status, p53, and p21 status [23], and the levels of apoptosis regulating B-cell lymphoma (Bcl) family of proteins [2,23,29,30], particularly the Bcl2/Bcl-2-associated X protein (Bax) ratio [23]. The cytotoxic effects of 2-DG are found to be higher under hypoxic conditions and the knockdown of HIF-1 significantly enhances the sensitivity of cells under hypoxia to 2-DG, suggesting that inhibition of HIF-1 may improve the clinical efficacy of glycolytic inhibitors such as 2-DG [14,16,23]. 2-DG has been found to be more toxic to tumor cells grown as spheroids (which develop microregions of hypoxia) when compared to monolayer cultures (MLCs) [14,23].
Cell death, induced by 2-DG, could be either apoptotic or necrotic depending on the cell type and environmental factors. While the induction of apoptosis has been found in c-Myc overexpressed cells, enhanced apoptotic death has been reported in drug-resistant human carcinoma cells (KB-DR) that could be linked to overexpression of glut receptors induced by 2-DG [23]. It has been shown that induction of apoptosis by 2-DG has been independent of Bcl2, and cytotoxic effects of 2-DG do not correlate with p53 status [23]. Susceptibility of p53 overexpressing cells to 2-DG is reduced by higher levels of catalase or glutathione peroxidase suggesting that the mechanism underlying enhanced cell killing by 2-DG in p53 overexpressing cells involves oxidative stress. Glucose and oxygen are potent regulators of glycolytic enzyme gene transcripts and, therefore, genetic alterations other than c-Myc activation are also expected to sensitize transformed cells to glucose deprivation. Furthermore, glucose by itself stimulates transcription of gene encoding glycolytic enzymes through the carbohydrate response element (choRE), a CACGTG motif, which has the same sequence as the core binding site for c-Myc [23].
2-DEOXY-D-GLUCOSE AS RADIOSENSITATOR
Analyzing the radiomodifying effects of 2-DG observed in several tumor cell lines reveal that the time of administration of 2-DG with respect to irradiation plays a critical role in determining the effects. Sensitization is generally found to be higher when 2-DG is added either just before (<5 minutes) or immediately after (<5 minutes) irradiation [10,14,23], is present in the incubating medium for 2 to 4 hours [14,17,23]. It has also been shown that the presence of the glycolytic inhibitor, 2-DG, for a few hours after irradiation can selectively inhibit the post irradiation repair processes in cells with high rates of glycolysis, such as cancer cells, thereby enhancing the damage caused by radiation [15,16,28].
Alterations in the expression of many genes involved in damage response pathway including DNA repair and apoptosis, transcriptional regulators, cell signaling, besides energy metabolism have been reported, which can significantly influence the radiosensitization of tumor cells. A great degree of heterogeneity in the 2-DG-induced modifications in radiation responses has been observed among the various human tumor cell lines that does not correlate well with the extent of decrease in the energy status (ATP levels), suggesting thereby that other disturbances caused by 2-DG also play important roles in the modifications of cellular responses to damage caused by radiation and chemotherapeutic drugs. These include (but are not restricted to) level of glucose transporters (GLUT1 and GLUT2), prosurvival and prodeath regulators, namely, c-Myc, ras, p53, p21, Bcl2/Bax ratio, and so on, and imbalances in the oxidative stress [23].
The degree of radiosensitization by 2-DG in multicellular tumor spheroids (MTS) generated from human glioma cell line (BMG-1) was found to be nearly 2.5-fold higher than in the MLCs, which correlated with the enhanced glycolysis in MTS [23], and with the role of synergy between endogenous oxidative stress related to tumors and induced metabolic oxidative stress [14].
Many studies suggested that 2-DG enhances the damage caused by chemotherapeutic drugs and ionizing radiation selectively in cancer cells while reducing the damage to normal cells [14,21,23]. 2-DG sensitizes cancer cells to radiation through mechanisms such as inhibiting DNA repair processes and recovery from potentially lethal damage [21,23]. However, despite of several past attempts to test the role of 2-DG in radiation therapy, only one clinical trial study on human cerebral gliomas has demonstrated that 2-DG improves the efficacy of radiotherapy [10,15,21].
Radiosensitization has also been suggested to be due to disruption of thiol metabolism resulting in oxidative stress-related cell death in the form of apoptosis [23]. It is pertinent to mention here that an inappropriate design of protocols may in fact reduce the efficacy of primary therapeutic agents by 2-DG as has been reported for a combination of radioimmunotherapies [23]. Treatment of human breast cancer cell lines with 2-DG results in the cessation of cell growth in a dose dependent manner [17]. However, evaluation of glucose usage, lactate production, and energy status could be useful for predicting the responses of tumors to the combined treatment of radiation and 2-DG [10].
2-DEOXY-D-GLUCOSE AS A CHEMOTHERAPY DRUG
2-DG acts synergistically with specific chemotherapeutic agents in causing cell death, and the class of chemicals that are most sensitive appears to be those that cause DNA damage [19-21,23]. Furthermore, 2-DG has been shown to inhibit the transcription of human papilloma virus, suggesting it to be an ideal adjuvant for enhancing the efficacy of chemotherapy in the treatment of drug-resistant cervical cancers [23]. 2-DG also enhanced the cytotoxicity of cisplatin and doxorubicin [16,20,23,27]. According to the results of some studies [20,23,27,31], it has been proposed that 2-DG may be a good chemosensitizer for chemoresistant patients as it alters reactive oxygen species (ROS) or redox state and sensitizes the cells to further damage caused by chemo agents. It was found that the combination of 2-DG and doxorubicin have a significant cell killing capability in rapidly dividing cells (such as T47D breast cancer cell line) compared with 2-DG or doxorubicin alone, whereas no effect was seen in slowly growing cells (such as MCF-7 breast cancer cell line) [19]. 2-DG has shown promising results as an adjuvant of radiation therapy and chemotherapy both in vitro and in vivo [14].
NORMAL TISSUE TOXICITY
In normal cells, growth is regulated by external growth signals and nutrient support. Cancer cells, in contrast, have lost responsiveness to most external growth signal, and as a consequence, nutrient supply in the form of glucose likely plays a unique role in maintaining cancer cell viability. Thus, normal and transformed cells respond to nutrient depletion or glucose deprivation in opposing manners. Whereas normal cells compensate by increased glucose transporter expression or modification, transformed cells are stressed by glucose-deprivation leading to the expression of an array of stress related genes, which is subsequently followed by cell death. In many normal cell types, glucose deprivation results in an increase in the maximum velocity of glucose transport. This has been attributed to one of several mechanisms; translocation of transporter from an intracellular compartment to the plasma membrane, changes in the glycosylation pattern of the Glut1 transporter with decreased turnover of the protein, or by increased synthesis of mRNA and protein [17]. When glycolysis is inhibited, the intact mitochondria in normal cells enable them to use alternative energy sources such as fatty acids and amino acids to produce metabolic intermediates channeled to the tricarboxylic acid cycle for ATP production through respiration [16,18]. As such, cells with normal mitochondria are expected to be less sensitive to agents that inhibit glycolysis [16].
2-DG produced a four to five fold greater effect in anaerobically growing cells than in aerobically growing cells. Consequences of glycolysis blocking is different in aerobic versus hypoxic cells. In the aerobic cell, if glycolysis is inhibited by 2-DG, ATP cannot be generated by this pathway. However, since O2 is available to the mitochondria, amino and/or fatty acids can act as energy-providing carbon sources for oxidative phosphorylation (OxPhos) to take place, producing ATP. In contrast, when glycolysis is blocked in the hypoxic cell the other carbon sources cannot be used by mitochondria as O2 is unavailable and consequently OxPhos cannot take place. Thus, when glycolysis is blocked in the hypoxic cell, it has no alternative means for generating ATP and, therefore, will eventually succumb to this treatment [18].
DISCUSSION
In general, cancer cells exhibit increased glycolysis and pentose-phosphate cycle activity, while demonstrating only slightly reduced rates of respiration. Initially these metabolic differences were thought to arise as a result of "damage" to the respiratory mechanism, and tumor cells were thought to compensate for this defect by increasing glycolysis. However, if cancer cells increase glucose metabolism to form pyruvate and nicotinamide adenine dinucleotide phosphate (NADPH) as a compensatory mechanism, in response to ROS formed as byproducts of oxidative energy metabolism, then inhibition of glucose metabolism would be expected to sensitize cancer cells to agents that increase levels of hydroperoxides (i.e., ionizing radiation and chemotherapy agents such as quinones that are known to the redox cycle and produce ROS). Although it is not possible to deprive cells of glucose in vivo, it is possible to treat tumor-bearing animals and humans with 2-DG, a relatively non-toxic analogy of glucose that competes with glucose for uptake via the glucose transporters as well as being phosphorylated by hexokinase at the entry point to glycolysis. Competition between 2-DG and glucose is thought to cause inhibition of glucose metabolism, thereby creating a chemically induced state of glucose deprivation. Although there are reports that the phosphorylated form of 2-DG (2-DG-6-P) can proceed through the first step in the pentose cycle (glucose-6-phosphate dehydrogenase) leading to the regeneration of one molecule of NADPH, 2-DG-6-P appears to be incapable of further metabolizing in the pentose cycle as well as incapable of metabolism to pyruvate. Ahmad et al. [20] have shown that administration of 2-DG to mice could be an effective way to inhibit glucose metabolism without causing toxicity until very high levels are achieved (lethal dose 50 ≥2 g/kg body weight) and could be tolerable in humans when administered up to 200 mg/kg. Therefore, using 2-DG as an inhibitor of glucose metabolism in vivo may provide a very effective addition to multi modality cancer therapies designed to limit hydroperoxide metabolism for the purpose of enhancing radio- and chemosensitivity in human cancers.
Singh et al. [32] have shown that the growth rate of rapidly dividing DU145 prostate cancer cells depend on high levels of glucose consumption, whereas the growth rate of relatively slow-growing LNCaP cells are much less dependent on glucose. They found a direct correlation between glycolytic capacity and degree of growth inhibition in response to glucose deprivation for these two cell lines. Their results are also consistent with earlier studies that showed a direct relationship between glycolytic capacity and growth rate for several rat hepatoma tumors, and lend further support to the hypothesis that high glucose consumption is required for rapid proliferation of most cancer cells. Zhao et al. [33] have also showed that increased aerobic glycolysis is a hallmark of cancer and that inhibition of glycolysis may offer a promising strategy to preferentially kill cancer cells. They proposed that trastuzumab to have remarkable efficacy in treatment of avian erythroblastosis oncogene B2 (ErbB2)-positive breast cancers when used alone or in combination with other chemotherapeutics. In their study, it is suggested that trastuzumab has antitumor effects in combination with glycolysis inhibitors in ErbB2-positive breast cancer by inhibiting glycolysis via downregulation of heat shock factor 1 (HSF1) and lactate dehydrogenase A (LDHA) in ErbB2-positive cancer cells, resulting in tumor growth inhibition. Moreover, increased glycolysis via HSF1 and LDHA contributes to trastuzumab resistance [32]. It is understood that glucose metabolism appears to be involved in the detoxification of intracellular hydroperoxides, and other authors have suggested that tumor cells demonstrate increased intracellular hydroperoxide production. It is proposed that the extent to which tumor cells increase their metabolism of glucose is predictive of tumor susceptibility to glucose deprivation induced cytotoxicity and oxidative stress. Therefore, when deprived of glucose using inhibitors of glycolytic metabolism (i.e., 2-DG), tumor cells with high glucose utilization will be more sensitive to cell death resulting from respiratory dependent metabolic oxidative stress than tumor cells with low glucose utilization and normal cells. It was hypothesized that the reason for this is because cancer cells with high glucose utilization generate more O2 and H2O2 from their mitochondrial electron transport chains. 2-DG is clinically a relevant competitor for glucose, thereby creating a chemically induced state of glucose deprivation. 2-DG inhibits glucose metabolism in animals, and it is not toxic to them except at very high levels (>2 g/kg body weight). It is tolerable in humans up to 200 mg/kg of body weight [19].
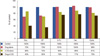
Zhang and Aft [21], on analyzing the effect of 2-DG with/without some other chemodrugs (Figure 3), have reported that there was a greater additive effect on cell cytotoxicity of 2-DG in combination with doxorubicin, 5-fluorouracil, trastuzumab, and cyclophosphamide. On one hand, they did not observe enhanced cytotoxicity of 2-DG with cisplatin in breast cancer cells, which exists in head and neck cancers [20]. On the other hand, in vivo obtained data have shown radio-sensitization effects of 2-DG. In this study, treatment with 2-DG or with radiation significantly inhibits tumor growth compared to the control group. While high doses of radiation almost completely suppressed tumor growth in the radiation-treated group, addition of 2-DG further enhanced the efficacy of radiation. More importantly, the radiation enhancement effect is stable long after the combined treatments of 2-DG and radiation. Some previous studies also showed that 2-DG increases the effect of radiation in pancreatic cancer and in head and neck cancers in mice. Thus, with p53-positive cancer, a lower dose of radiation could be effective when used in combination with 2-DG [21,31].
CONCLUSION
The precise molecular mechanisms underlying the cellular responses to metabolic stress induced by 2-DG alone and in combination with other cytotoxic agents such as ionizing radiation and chemotherapeutic agents appear to be complex and remain to be completely elucidated. Elucidation of various mechanisms underlying radiosensitization and chemosensitization by 2-DG using established tumor cell lines will be very useful in designing effective protocols using 2-DG in cancer therapy.

 XML Download
XML Download