

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
In recent years, significant advances in the understanding of the molecular biology in breast cancer development have led to the identification of new molecular target and the development of targeted therapy. Much more efforts are also being made in the development of better molecular targets which are crucial for tumorigenesis and metastasis. In this context, heat shock proteins (HSPs) has gained interest as a promising anticancer drug target due to its involvement at the crossroads of multiple signaling pathways associated with cell proliferation and cell survival [1-3].
HSPs were first discovered in Drosophilia melanogaster in 1962 as a set of proteins that was rapidly induced in response to thermal stress [4]. Thereafter, many studies demonstrated that HSPs are a highly conserved family of proteins either expressed constitutively or regulated inductively by various cellular stresses such as inflammation, toxins, hypoxia, and radiation in all living organisms [5,6].
HSPs are highly abundant proteins in eukaryotic cells, constituting about 1-2% of total proteins in unstressed cells and increasing to 4-6% of cellular proteins under stress [7,8]. Under stress conditions, HSPs are rapidly induced through transcription and translation mechanisms. The transcription of HSP genes is regulated by a family of heat shock transcription factors (HSFs). The HSF family includes HSF1, main regulator of the short-term induction of HSPs. Under unstressed conditions, HSF1 exists as a inactive cytosolic monomer, bound to HSPs. However, in stressed cell, HSF1 dissociates from HSPs and is transported to the nucleus where it subsequently forms phosphorylated homotrimer. Then it binds to the promoter site of HSPs gene, leading to HSPs production. If HSPs are over-expressed in the absence of stress it binds directly to the HSF1 trans-activation domain resulting in its suppression [9,10]. Interestingly, HSF1 has been proposed to affect tumor initiation and progression. Recent reports demonstrated that HSF1 plays a key role in the development of tumors associated with activation of Ras or inactivation of p53 and HSF1 inactivation inhibits the progression of a wide spectrum of cancers [11,12]. Meng et al. [13] reported HSF1 is critical for proliferation of HER2-expressing cells, most likely because it maintains the level of HSPs, which in turn control regulators of senescence p21 and survivin.
HSPs form multimolecular complexes and act as molecular chaperones, binding other proteins named client proteins. Their principal function as chaperones is the maintenance of protein stability under normal conditions and prevention of stress-induced cellular damage which can be accomplished in several ways, including protein folding, prevention of protein aggregation, stability or proteasomal degradation of selected proteins and transport of proteins [14-16]. Most HSPs are also known to play an important and complex role in apoptosis, interacting with components of apoptosis pathway or activating antiapoptotic mediators [17,18]. Mammalian HSPs have been classified into five main families according to their molecular weight: HSP100, HSP90, HSP70, HSP60 and small HSPs (15-30 kDa) including HSP27 [14,19]. High molecular weight HSPs are ATP-dependent chaperones, while small HSPs act in an ATP-independent manner [20,21].
Interestingly, recent data showed essential roles of HSPs in malignant process. Expression of high levels of HSPs has been observed in a wide range of human cancers including gastric, breast, endometrial, ovarian, colon, lung, and prostate [22]. The expression of several HSPs has also been shown to be correlated with tumor cell proliferation, differentiation and apoptosis in several types of cancer. More specifically, high expression of HSP90 and HSP70 has been correlated with poor prognosis in breast cancer [23].
This article reviews the physical roles of HSPs in malignant cell, especially breast cancer and the mechanisms by which inhibition of HSPs may be useful in targeted cancer therapy.
THE FAMILY OF HSPs IN CANCER
Small HSPs (HSP27)
HSP27 is a member of the small HSPs family that acts as an ATP-independent chaperone and mainly localized in the cytosol. They are potent mediator of protein folding and also involved in architecture of cytoskeleton, cell migration, cell growth/differentiation, and tumor progression [15,21,24,25]. HSP27 also has antiapoptotic property [26].
High levels of HSP27 have been observed in many cancer cells including breast carcinoma [27,28], compared to normal cells in which expression is undetectable or relatively low [15]. Moreover, its aberrant expression in cancer is associated with aggressive tumor behavior, increased resistance to chemotherapy, and poor prognosis for the patients.
HSP27 is activated in various stress conditions both by transcriptional activation and posttranslational modification (phosphorylation). HSP27 can be phosphorylated at three serine residues 15, 78, and 82, and its phosphorylation is mediated by the p38 MAPK stress kinase pathway [29]. This phosphorylation is a reversible event that modulates the oligomerization of HSP27. Different cellular functions of HSP27 seem to be related to its oligomerization state. Moreover, various different phosphorylation patterns of HSP27 have been found to be associated with the aggressiveness of different tumor types. A recent report demonstrated a twofold increased phophorylation of HSP27 at serine 78, but not serine 15 and 82, in HER2 positive breast cancer samples compared to HER2 negative tumors. However, the exact role of HSP27 phosphorylation in the physiology of cancer remains incompletely understood.
Recently, several studies demonstrated that the overexpression of HSP27 seems to be correlated with increased resistance to chemotherapeutic drug-induced apoptosis in cancer cells [30,31]. Hansen et al. [32] reported the inhibition of doxorubicin induced apoptosis in the HSP27 overexpressing breast cancer cell, demonstrating a protective role of HSP27 against apoptosis. In addition, a recent report presented that upregulation of HSP27 in breast cancer cells reduces trastuzumab susceptibility by increasing HER2 protein stability [33]. These recent studies suggest possibility of HSP27 inhibition as molecular target for cancer therapy. However, unlike other HSPs, the small HSPs do not bind ATP, and it makes this molecule problematic for targeting with small compounds.
HSP70
Human cells contain several HSP70 family members including the stress-inducible HSP70 (also called HSP72 or HSPA1) and the constitutively expressed heat shock cognate 70 (HSC70, HSP73 or HSPA8) in the cytosol and nucleus, mitochondrial HSP70 (Grp75, Mortalin or HSPA9), and glucose regulated protein 78 (Grp78, HSPA5) in the endoplasmic reticulum [34-36]. HSP70, like other HSPs, is a molecular chaperone expressed in response to stress. Under normal conditions, HSP70 also plays multiple roles, including the folding of newly synthesized proteins [37,38], the transport of proteins and vesicles [39], the assembly and dissociation of multi-protein complexes [40], and the degradation of denature proteins [41,42].
HSP70 is also powerful anti-apoptotic protein that acts at different key points, affecting both the extrinsic and intrinsic pathway of apoptosis. For example, HSP70 was reported to inhibit the important apoptotic mediator, Bax translocation, thus preventing mitochondrial membrane permeabilization. Together with its co-chaperone HSP40, HSP70 also blocks TNF-induced apoptosis. Moreover, HSP70 directly interacts with apoptosis protease activating factor-1 (Apaf-1), thereby inhibiting recruitment of procaspase-9 to the apoptosome and the consequent caspase-3 activation. HSP70 can also block caspase-independent signaling through inhibition of apoptosis-inducing factor (AIF)-induced chromatin condensation and cathepsins release. In conclusion, HSP70 regulates apoptosis by inhibiting stress-induced signals, by preventing mitochondrial membrane permeabilization, and by suppressing caspase activation and DNA fragmentation [26,43]. HSP70 also plays role in senescence through effects on the p53-p21 pathway [44].
HSP70 is expressed at high levels in a wide spectrum of cancer cells and HSP 70 expression has been routinely associated with poor prognosis [43]. The exact role of the HSP70 in cancer remains to be elucidated. However, in cancer cell, HSP70 overexpression is thought to provide a survival advantage due to its ability to inhibit apoptosis and senescence [26,45]. Moreover, the HSP70 also acts as co-chaperones for HSP90 by its essential role in the substrate-loading phase of the HSP90 molecular chaperone cycle, key to the stability and function of multiple oncoproteins.
Many studies reported that HSP70 overexpression has also been correlated with therapeutic resistance [46]. Although the detailed mechanisms of resistance remain to be elucidated, recent evidence suggests that reduced activation of ERK, NF-κB, and JNK pathways may be responsible [47]. Moreover, pharmacological inhibition of HSP90 has been found to induce a compensatory expression of HSP70 [48]. This might be because HSP70 is a highly protective protein that may strongly reduce the cell death effect induced by HSP90 inhibition. In this view, the potential therapeutic benefit of modulating HSP70 activity has become attractive, especially dual therapy against both HSP90 and HSP90 [49]. The search for inhibitors of HSP70 has dramatically increased over the last 2 years; however, there are limited small molecule inhibitors of HSP 70 available.
HSP90
HSP90 is a highly abundant and evolutionarily conserved protein in the all eukaryotic cell. Five HSP90 isoforms have been identified to date, including the two major cytoplasmic isoforms, HPS90α and HPS90β (also called HSPC1 and HSPC3, respectively), endoplasmic reticulum localized glucose regulated protein 94 (Grp94), mitochondrial tumor necrosis factor receptor-associated protein 1 (TRAP1), and membrane-associated HSP90N [16]. Despite their different cellular localization, these isoforms have a similar overall structure and function as chaperone by a common mechanism involving the cyclic conformational change.
HSP90 exists as a homodimer within the cell, and each subunit is composed of three domains: N-terminal ATPase domain, middle domain implicated in client protein binding, and C-terminal domain containing protein-protein interaction and dimerization motif [50]. The N-terminal domain contains ATP-binding pocket, and the chaperoning activity of HSP90 requires both the binding and hydrolysis of ATP at this site [51]. Besides the role of C-terminal domain in dimerization, it was suggested that the C-terminal domain contains a second ATP-binding site of HSP90 [52-54]. The contribution of this second site to the overall regulation of the chaperone is still unknown. The C-terminal domain also recruits co-chaperones through a conserved tetratricopeptide repeat (TPR)-binding motif, EEVD [55]. Co-chaperones, containing TPR domains such as HOP, and the non-TPR co-chaperones, CDC37, p23, and Aha1, play an important role in client protein maturation and modulation of ATPase activity [39,19]. Some of these co-chaperones such as Aha1 and HSP70 have been proposed for independent molecular targets [56].
HSP90 functions as a part of a multichaperone complex via association with a variety co-chaperons and client proteins that rely on the complex for maturation and stability. The HSP90 complex appears to cycle between an ADP-bound and ATP-bound state [57]. In an ATP-bound state, HSP90 undergoes a conformational change and becomes a mature complex that is essential for it to perform its function of client protein folding and stabilization. The hydrolysis of ATP to ADP facilitates release of these client proteins, and then they are degraded by ubiquitin proteasome pathway [58].
HSP90 is important molecular chaperone that regulates the stability and activity of numerous client proteins covering almost all cellular processes. More than 200 client proteins have been identified so far, and the list is constantly being updated [59]. Its client proteins include BCL-ABL, SRC, HER2, EGFR, CRAF, BRAF, AKT, MET, VEGFR, FLT3, androgen and estrogen receptor, hypoxia-inducible factor (HIF)-1α, and telomerase that are directly involved in malignancy and mutated oncogenic proteins that are required for the transformed phenotype. These include proteins important in breast cancer progression such as HER2 and c-SRC. Indeed, HSP90 overexpression has been observed in a variety of human malignancies including the breast cancer [59].
There are several principal functions of HSP90 in malignant cells. As mentioned above, HSP90 stabilizes many oncogenic proteins in cancer cell. HSP90 may inhibit apoptosis through several interactions. For example, it has been reported that HSP90 binds directly to apoptotic protease activating factor 1 (Apaf-1), and inhibits its oligomerization, recruitment of procaspase-9, thus blocking the assembly of a apoptosome [60]. Moreover, increased expression of HSP90 has been implicated in resistance to senescence due to its essential role of telomerase stability [61]. HSP90 also has a role in angiogenesis owing to its stabilizing properties on the transcription factor HIF-1α, and VEGF and nitric oxide synthase, two basic players in angiogenesis, are HSP90 client proteins. Finally, HSP90 may play a role in tumor invasion and metastasis. Interestingly, HSP90 inhibitors have been implicated in bone metastasis in breast cancer, but its mechanism is not fully explained [62].
HSPs INHIBITION AS A THERAPEUTIC STRATEGY
The cytoprotective function of HSPs is essential for cancer cell survival and high expression of HSPs is correlated with a poor clinical outcome. HSPs are an attractive and interesting molecular target in cancer therapy, particularly HSP90 that controls many oncoproteins and different signaling pathways in cancer cell. Many inhibitors of HSP90 have been developed and undergone clinical trials. While HSP27 and HSP70 are undoubtedly implicated as potential target for anti-cancer therapy, their clinical evaluation has not started yet.
Targeting HSP90 in breast cancer
Many kinds of HSP90 inhibitors have been identified so far. The majority of HSP90 inhibitors bind to the N-terminal ATP-binding site of HSP90 and inhibit the ATPase cycle which is essential for the HSP90 chaperone activity [48]. Therefore, HSP90 inhibition results in degradation of important oncogenic client proteins by the ubiquitin-proteasome pathway, inhibition of tumor growth and activation of apoptosis in cancer cells. Although blockade of N-terminal ATP binding of HSP90 has been focus of drug development, distinct modes of inhibition are being considered and include disruption of co-chaperone-HSP90 interactions, inhibition of the C-terminal ATP binding site, or inhibition of client-HSP90 interaction.
The natural products, geldanamycin (GA) and radicicol were first inhibitors discovered. Geldanamycin, an ansamycin antibiotic, was first isolated from Streptomyces hygroscopicus and noted to have inhibitory activity against HSP90 [63,64]. Undesirable properties associated with GA, such as hepatotoxicity and poor solubility [65], led to a necessary round of compound optimization. Therefore, the less toxic and more effective GA derivatives, 17-allylamino-17-demethogeldanamycin (17-AAG, tanespimycin, KOS-953), 17-dimethylaminoethylamino-17-demethoxygeldanamycin hydrochloride (17-DMAG, alvespimycin, KOS-1022), and IPI-504 (retaspimycin) have been developed as potential therapeutics in a variety of clinical trials [66]. More recently, purine-scaffold derivates such as PU-H71, PU-DZ8, and CNF2024 (BIIB021) developed based on the structure of the nucleotide ligand [67,68]. Because of the potential toxicity of GA derivatives, specific small molecular weight HSP90 inhibitors may be more effective clinical agents. Several small molecular weight HSP90 inhibitors, including SNX-5422, CNF2024, STA 9090, and AUY 922, are currently in clinical trials in various tumor types [69-72]. Current phase I and II clinical trials with HSP90 inhibitors in breast cancer are seen in Table 1. Interestingly, HSP90 isolated from tumor cells has a binding affinity for the inhibitors between 20 and 200 times higher than does HSP90 isolated from normal cells. This might be due to the fact that tumor cells, as compared to their counterparts, might exhibit a stressed phenotype, with an enhanced dependency on the cytoprotective action of HSP90. This 'addiction' of cancer cells to HSP 90 client proteins have been proposed as rationale for selectivity of HSP90 inhibitors for cancer versus normal cells [73].
Breast cancer is good target of HSP90 inhibitor for several reasons. HER2 is among the most sensitive client proteins of HSP90, demonstrating degradation within 2 hours of HSP90 inhibition in cell culture experiments [74] and HSP90 inhibitors have shown activity in HER2-deriven xenograft model [75]. Modulation of estrogen and progesterone receptor has been long-standing target of breast cancer and both receptors are also client proteins of HSP90. Moreover, resistance of breast cancer cells to chemotherapy is known to involve the phosphatidylinositol 3-kinase (PI3K) pathway [76], and its key signaling protein Akt is modulated by HSP90. In addition, the expression of HSP90 has been shown to be correlated to adverse clinical prognosis.
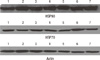
We previously investigated the HSP70/90 expression and the effect of HSPs inhibitor in breast cancer [77]. We found that more prominent HSP90 expression in breast cancer tissue than in benign tissue by immunohistochemistry staining and it was related to more aggressive tumor type; positive lymph node status and poor differentiated histologic type. However, we could not find the difference of HSP70 expression between two lesions (Table 2). We also demonstrated the expression of HSP70/90 in breast cancer cell lines using the Western blot (Figure 1). In this study, we investigated the effect of HSP90 inhibitor (GA) on cell growth of the human breast cancer cell lines: MDA-MB231, MDA-MB 435, MCF-7, and T47D cell line. GA markedly inhibited the cell growth of these cell lines in a dose-dependent manner (Figure 2).
HER2 overexpression is observed in 20-25% of breast cancer patients and it predicts for a poor clinical outcome. HSP90 expression is associated with HER2 expression [78]. Preclinical studies have demonstrated the notable sensitivity of HER2-overexpressing breast tumors to HSP90 inhibitors [69,70]. 17-AAG (tanespimycin) is being developed in the clinic for HER2 positive breast cancer and demonstrates a moderate clinical response in combination with trastuzumab in patients with trastuzumab-refractory HER2-positive metastatic breast cancer in recent report [79]. In phase II study, 31 patients (metastatic HER2-positive breast cancer progressing on trastuzumab) received weekly tanespimycin at 450 mg/m2 intravenously and trastuzumab. The most common toxicities were diarrhea, fatigue, nausea, and headache. The overall response rate was 22% and the clinical benefit including complete response, partial response, and stable disease was 59%. IPI-504 (retaspimycin), a 17-AAG analogue, has improved water solubility properties thereby facilitating formulation for parental administration. In breast cancer, Phase I/II trials are currently underway to evaluate the dosing schedules [80]. Moreover, a HSP90 inhibitor (SNX-5422) inhibits p95-HER2, the truncated form of HER2 associated with trastuzumab resistance, and suppress their growth [81]. Similarly, 17-DMAG can overcome resistance of HER2 positive breast cancer cells to aromatase inhibitors [82].
Triple negative breast cancer (TNBC; defined by the lack of expression of estrogen, progesterone, and HER2) patients have poor prognosis and survival outcomes, but there are currently no specific targeted therapies. Clinical studies have been shown the EGFR overexpression and activation of PI3K pathway in TNBCs and it has been associated with poor prognosis. Hence, HSP90 inhibitors may provide an opportunity to inhibit tumor progression of TNBCs because the many of HSP90 client proteins are oncoproteins including EGFR and involved in multiple oncogenic signaling pathways. Interestingly, PU-H71 (purine based synthetic HSP90 inhibitor) induces tumor regression in a xenograft model of TNBCs and that are not candidate for 17-AAG treatment [83].
CONCLUSIONS
HSPs are highly expressed in many malignant human tumors including breast cancer, and the cytoprotective chaperone function of HSPs is essential for cancer cell survival. Moreover, these proteins seem to be associated with a poor clinical outcome and poor response to therapy. As a consequence, HSPs is an exciting new target in cancer therapy, particularly HSP90 that modulates multiple oncogenic proteins and signaling pathways in cancer cells. Clinical activity has been seen with HSP90 inhibitors like 17-AAG and 17-DMAG in breast cancer, especially trastuzumab-resistant cancer and many clinical trials are currently underway. The results of clinical phase II and III trials evaluating the efficacy of these drugs are awaited.
Also, although not described in this review, another potential of HSPs as vaccine properties should be mentioned. Concerning extracellular HSPs, they can act as chaperones for tumor peptide antigens thereby eliciting an immune anti-tumor response. Hence, HSPs can be used for vaccine preparations and this approach adds to the overall interest of HSPs in cancer therapy.



 XML Download
XML Download