

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Breast cancer has become the most common cancer in women, constituting 23% of all cancers, with over one million new cases diagnosed every year worldwide.(1) Breast cancer mortality has either been stable or has decreased in most countries because of earlier diagnosis and improvements in treatment. Breast cancer treatment has recently shown great progress, and adjuvant chemotherapy has been supportively employed in many patients. Anthracycline-based regimens are the first-choice of practitioners for treatment of breast cancer. Taxane may become the drug choice for adjuvant chemotherapy, perhaps in combination with anthracycline. More recently, targeted therapeutics, such as trastuzumab, lapatinib, and bevacizumab, have been approved for treatment of breast cancer.(2)
DNA alterations resulting from genetic and epigenetic events have been implicated in cancer onset and progression. Epigenetic events are a major cause of the transcriptional repression of tumor suppressor gene in the absence of DNA mutations.(3) The two main epigenetic mechanisms are DNA methylation and histone modification. Histone acetylation is regulated the opposing actions of histone acetyltransferases (HATs) and histone deacetylase (HDAC). Histone acetylation by HATs masks positive charges of lysine residues on histone tails, thus reducing tight interactions between histones and negatively charged DNA. In contrast, histone deacetylation allows histones to wrap more tightly around DNA, thus limiting accessibility to DNA for transcription.(4,5) HDAC inhibitors (HDACIs) cause accumulation of acetylated histones in nucleosomes, resulting in increased gene transcription which has been shown to result in growth arrest, differentiation, and apoptosis of several types of cancer cells in vitro and They are known to have anticancer activities in animal models.(5)
Several structural classes of HDACI have been developed, These include short-chain fatty acids (such as valproic acid and butylate), cyclic tetrapeptides (e.g., FK228), hydroxamic acids (such as TSA, vorinostat [SAHA], LBH-589, LAQ824 and belinostat [PXD101]) and amides (e.g., MS-275 and MGCD0103).(5) SAHA, PXD101, LBH589, and LAQ824 have been useful as mono-therapies in cutaneous T-cell lymphoma patients. Early-phase clinical trials on HDACIs are currently underway to evaluate the therapeutic efficacies of these compounds against solid tumors and hematological malignancies.(6) Preclinical studies have shown that SAHA was associated with differentiation and apoptosis of breast cancer cells, causing accumulation of cells in G1, and then next in the G2-M phase, of the cell cycle, giving rise to morphological changes including flattening and enlargement of the cytoplasm as well as a decrease in the nuclear-cytoplasmic ratio.(7) A Phase II study of the single-agent SAHA has been conducted in patients with metastatic breast cancer. The tolerable toxicities observed, and the potential for clinical benefit, suggest that further assessment of SAHA as part of a combination therapy with either chemotherapeutic or targeted agents in metastatic breast cancer patients might be of value.(8)
Cancers are highly individual in responses to chemotherapy, and chemosensitivity assays are thus of great value to patients receiving chemotherapy. The histoculture drug response assay (HDRA), the ATP-based chemotherapy response assay, the microphysiometer bioassay, and the drug-sensitivity pattern analysis are all employed for in vitro drug sensitivity tests. Among these assay, HDRA is a three-dimensional histoculture method using a collagen gel in combination with the MTT [3-(4,5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide] cell death assay. In the HDRA, approximately 1 mm3 of cancer tissue is cultured on a collagen gel without enzymatic tissue digestion. Cell to cell contact is maintained and long-term culture is possible under conditions resembling those in vivo, allowing cancer cells to show high survival rates even days after exposure to anticancer drugs.(9) Such three-dimensional tissue culture permits maintenance of intercellular contacts and cancer cell interactions with stromal cells unlike the usual two-dimensional monolayer culture, where a major limitation is a lack of stroma. It appears that HDRA can evaluate the sensitivities of tumor cells to anticancer drugs under conditions similar to those in vivo and, therefore, the in vivo drug sensitivity show a high prediction rate.(10) Chemosensitivity data from HDRA correlated with clinical outcomes after chemotherapy, and the HDRA was useful in selection of effective drugs.(11-13)
In this study, we assess the efficacy of HDACIs (one novel and two commercially available compounds) on breast cancer cells, compared with other established drug regimens, using HDRA. Additionally, tumor responses were examined with reference to clinicopathological features, to identify individual candidate chemosensitivity markers.
METHODS
Patients
A total of 103 breast cancer patients who underwent mastectomy were prospectively enrolled in this trial. Eligibility criteria included pathologically proven breast cancer, an expectation of adjuvant chemotherapy, Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, and an age of 75 yr or less. Patients were excluded if they had received neoadjuvant chemotherapy. Among all patients, 25 were excluded because the tumor samples were inadequate. Baseline demographic and clinicopathologic characteristics of the 78 patients are shown in Table 1. All tumor samples were acquired after written informed consent was given, and the study was conducted with the approval of the Institutional Review Board (IRB No.2009-0091) for Human Genetic and Genomic Research, in accordance with the Declaration of Helsinki.
Anti-cancer drugs
The established drug regimens tested included doxorubicin, cyclophosphamide, a combination of doxorubicin and cyclophosphamide (AC), paclitaxel, docetaxel, and a combination of doxorubicin and docetaxel (AT). The three HDACIs used were hydroxamic acid derivatives, one was the FDA-licensed SAHA (suberoylanilide hydroxamic acid, Merck Co., Whitehouse Station, USA), another the patented PXD101 (CuraGen Co., Branford, USA), and the third a novel drug candidate (CG-2; Crystal Genomics Co., Seoul, Korea), with 'patent pending' status. The concentrations of the established drugs used in this study were 6 µg/mL doxorubicin, 20 µg/mL cyclophosphamide, 10 µg/mL paclitaxel and 50 µg/mL docetaxel, in line with values employed previously.(14-18) For HDACIs, HDAC activity assays were performed using HeLa cell extracts and the Fleur de Lys™ fluorescent assay system (BioMol International LP, Plymouth Meeting, USA). IC50 values which were determined at 50% dose-dependent reduction in HDAC activity of the HDACI drugs were 1-130 nM. The anti-proliferative effects of HDACIs on a human colon cancer cell line, HCT116, were measured using the MTS (Formazan) assay system (Promega, Madison, USA). EC50 values were determined by a 50% dose-dependent reduction in cell viability after 48 hr of constant exposure to HDACIs in complete growth medium. EC50 values were between 0.22 and 1.1 µM. The final concentration of HDACIs used was estimated at 4 µg/mL by selecting median values close to the clinical and preclinical doses with minimal toxicity in vivo.
Novel HDAC inhibitor
CG-2, a novel HDACI candidate (patent pending), is a hydroxamic acid similar to SAHA and PXD101, but has linkers and hydrophobic domains distinct from those of SAHA and PXD101. The novel compound was designed to optimize scaffold using proprietary technology, as previously described.(19) The efficacy of this compound was evaluated by an enzyme inhibition assay, anti-proliferation assay with various cancer cell lines, florescence activated cell sorting apoptosis and cell cycle analyses, and measurement of acetylated histone accumulation. In vitro ADME (absorption, distribution, metabolism, and excretion) studies along with in vivo PK studies have also been performed. Finally, this novel hydroxamic acid derivative was shown to have anti-tumor activity without severe adverse events in animal studies using xenograft tumor models.
Histoculture drug response assay (HDRA)
The HDRA was used for evaluation of chemosensitivities as described earlier in colorectal cancer samples.(19) Briefly, three sections of tumor tissue (each 0.5 cm in diameter) were freshly harvested from mastectomy specimens. Tumor samples were washed in Hanks' balanced salt solution (HBSS; GIBCO, Gaithersburg, USA) and further minced into pieces approximately 1 mm in diameter in Hank's solution on a clean bench in 96-well plates. Four replicates were run for all control and treatment groups. Aliquots of media (100 µL) containing anti-cancer drugs were poured directly into each well, followed by incubation at 37℃ in a humidified atmosphere of 95% air/5% CO2 for 3 days. After histoculture, 100 µL of 0.06% collagenase type I (Sigma, St. Louis, USA) in HBSS and 0.2% MTT (Sigman, St. Louis, USA) in phosphate buffered saline were added to each well and plates were incubated for an additional 4 hr at 37℃. The medium was removed, and 0.5 mL dimethyl sulfoxide (DMSO) was added for solubilizing MTT-formazan. Absorbance of the solution in each well was read at 540 nm using a microplate reader (VersaMax, Sunnyvale, USA). The inhibition rate (IR) was calculated as following equations: IR (%)=(1-mean absorbance per gram of tumor specimen in drug-treated wells/mean absorbance per gram of l tumor specimen in control wells)×100. In our study, the IR cut-off values for chemoresponsiveness were selected as equal to or more than 30% (IR30) and 40% (IR40).
Statistical analysis
Inhibition rates of the respective regimens were examined using repeated measure ANOVA. IR30 and IR40 values were compared by chi-square test using McNemar. Univariate analysis of clinicopathologic features associated with chemosensitivity (at IR30) was performed by binary logistic regression. The significance level was considered to be 5% for each analysis, and all calculations were performed using SPSS software (version 14.0; SPSS Inc., Chicago, USA).
RESULTS
Tumor growth inhibition rates (IR) of established regimens
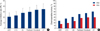
AT (combination of doxorubicin and docetaxel) showed the highest mean IR of an established drug regimen followed by docetaxel, paclitaxel, AC (combination of and cyclophosphamide), cyclophosphamide, and doxorubicin, in that order. Between-drug differences were statistically significant except for AC and paclitaxel, and docetaxel and AT (Figure 1A). The percentages of chemosensitive tumors (chemoresponsiveness) were 26.9-60.3% when the IR cut-off value was set at 30% (IR30), and 15.3-39.7% at a 40% cut-off (IR40) (Figure 1B). AT was the most effective regimen at IR30. The chemoresponsiveness of combined regimen (AC and AT) were higher than seen by any single agents (doxorubicin vs AT; p<0.001).
Tumor growth inhibition rates (IR) of HDACIs
The mean IR value of HDACIs was highest for PXD101, followed by SAHA and CG-2, in that order (Figure 2A). The IR values of PXD101 and SAHA were higher than that of the novel HDACI and significant differences were evident between PXD101 and CG-2 (p=0.032). Chemoresponsiveness were 73.1%, 73.1%, and 61.5% at IR30, but 57.7%, 46.7%, and 42.3% at IR40 for SAHA, PXD101, and CG-2, respectively (Figure 2B). However chemoresponsiveness differences at IR30 and IR40 were not statistically significant for any drug. The percentage of tumors chemoresponsive to HDACIs was evaluated among the tumors resistant to established drugs. In AC resistant tumors, the percentage of tumors chemoresponsive to SAHA, PXD101, and CG-2 were 38.5%, 44.9%, and 32.1%, respectively. For AT resistant tumors, chemoresponsive percentages were 23.1%, 23.1%, and 25.6% with SAHA, PXD101 and CG-2, respectively.
Relationships between clinicopathologic features and chemosensitivities
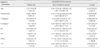
HER2/Neu overexpressing breast cancer cells were chemosensitive (at IR30) to SAHA and AT (p=0.031 and 0.042, respectively) (Table 2). AT was also effective against related to HER2/Neu overexpressing breast cancer cells (at IR40) (p=0.041). No further associations between drug sensitivities and clinicopathologic parameters were observed.
DISCUSSION
In HDRA, the three-dimensional tissue culture mode allows maintenance of intercellular contact and cancer cell interactions with stromal cells, unlike conventional two-dimensional monolayer culture, a major limitation in which there is a lack of stroma. Therefore, HDRA may be an appropriate method for evaluating drug sensitivity of breast cancer cells because the stoma of the mammary gland occupies over 80% of the resting breast volume. HDRA utilizes the MTT assay as a quantitative end-point. The MTT assay, which relies upon cellular metabolism of tetrazolium salts to yield strongly colored formazans, permits the evaluation of the total living metabolically active cell population, unlike clonogenic and radioactive assays that effectively evaluate only dividing cells.(20) MTT-based HDRA is a valuable tool in assessing total tumor cell viability, facilitating evaluation of both chemosensitivity and modes of interaction between established and novel drugs.(19) The overall and disease free-survival rates of patients presenting HDRA-sensitivity to drug have been found to be significantly higher than those of HDRA-resistant groups.(12,21) Chemoresponsiveness shown by HDRA had a 91.7% positive predictive value in breast cancer patients undergoing neoadjuvant chemotherapy.(22) In the present study, correlations between chemosensitivity and clinical outcomes of adjuvant chemotherapy were not evaluated because of short the follow-up times. Further study is necessary to assess the correlation of chemosensitivity shown by HDRA with clinical outcomes of adjuvant chemotherapy in breast cancer patients.
Conventional chemotherapeutic drugs are effective in killing proliferating cells but show reduced action against nonproliferating cells existing in the cores of solid tumors. This presents a problem with residual disease, in that quiescent cancer cells surviving a course of chemotherapy then subsequently present as a disease relapse. Chemotherapeutic drugs kill not only tumor cells but also proliferating stem cell populations in epithelial linings and the immune system, resulting in the many undesirable toxicities associated with chemotherapy. However, HDACIs are effective in killing both proliferating and nonproliferating tumor and immortalized cells, but neither proliferating nor arrested normal cells are affected.(23) HDACIs kill cells by either of two pathways, a rapid pathway involving cell transit through the defective G2 checkpoint, with resultant aberrant mitosis and rapid induction of apoptosis, or a slow pathway activated in G1 arrested cells, usually resulting in increased p21WAF1/CIP1 expression and retention of G1 arrest.(24)
The HDACIs used in this study were SAHA, PXD101, and CG-2 (a novel HDAC inhibitor). CG-2 is a hydroxamic acid derivative similar to SAHA and PXD101, but contains distinct linkers and hydrophobic domains. Because this novel compound has patents pending, the chemical structure is not discussed in the present study. The inhibition rate on tumor growth was highest when PXD101 was tested, followed by SAHA and CG2 (in that order). The HDACIs showed high chemosensitivities (>60%) at IR30. The mean IR value of the established drugs was highest for the AT combination, followed by docetaxel, paclitaxel, AC, cyclophosphamide, and doxorubicin (in that order), consistent with data from a previous study.(22)
Combinations of HDACI with established chemotherapeutics showed enhanced anti-tumor effects in both colorectal and breast cancer cell lines.(25,26) Because chromatin DNA is tightly compacted, inaccessibility of DNA to drugs targeting this material may reduce anti-cancer drug efficacies. Loosening of chromatin structure by histone acetylation may increase the efficiency of an established DNA-damaging anti-cancer drug. Trichostatin A (TSA) or SAHA first, followed by anticancer drugs may have an advantage for treating tumors resistant to these drug and increase the efficiency of the anti-cancer drug.(25,27) Both SAHA and PXD101 are currently in clinical trials in combinations with 5-FU because HDAC inhibition results in repression of thymidylate synthase in solid tumors.(19) Although chemosensitivities of combinations of HDACIs and established drugs were not investigated in this study, some tumors were chemoresponsive to HDACIs yet resistant to established drugs. In AC and AT resistant tumors, the percentages of HDACI chemoresponsive tumors were 32.1-44.9% and 23.1-25.6%, respectively. These results suggest that HDACIs can be active against established drug-resistant tumors and combinations of HDACIs with conventional drugs may yield additive effects. Further study is necessary to evaluate the sensitivities of HDACIs and established drug combinations, using HDRA of breast cancer tissues.
Approximately 25% of breast cancer patients show amplification and overexpression of the HER2/Neu oncogene, which encodes a member of the epidermal growth factor receptor tyrosine kinase family, promoting cell proliferation and survival. HER2/Neu overexpression has been associated with poor prognosis in breast cancer. HDACIs such as LAQ824 and SAHA up-regulated p21WAF1 and p27KIP1 increased the percentage of cells in the G2-M phase of the cell cycle, and induced apoptosis of HER2 overexpressing breast cancer cells. These agents induced acetylation of heat shock protein 90, leading to dissociation of HER2 from a chaperone molecule, promoting polyubiquitylation and degradation HER2. A combination of an HDACI with trastuzmab or docetaxel induced synergistic cytotoxic effects against HER2/Neu overexpressing breast cancer cells in vitro.(28,29) In the present work, HER2/Neu overexpressing breast cancer cells were chemosensitive (at IR30) to SAHA. This suggests that HER2/Neu overexpressing breast cancer cells are sensitive clinical targets for HDACIs.
CONCLUSION
The present work confirms the antitumor activities of HDACIs against breast cancer cells in vitro, supporting the use of HDACIs against molecular targets in anticancer therapy. The chemoresponsiveness of breast cancer cells to HDACIs, and the therapeutic efficacies noted, were comparable to data obtained using established drug regimens. HER2/Neu overexpressing breast cancer cells were sensitive to SAHA. In the absence of appropriate biomarkers identifying HDACI-responsive tumors, parameters predictive of tumor drug responses must be continuously sought.


 XML Download
XML Download