

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The estrogens, predominantly 17β-estradiol (E2), promote the growth and differentiation of sexual organs and other tissues related to reproduction. While estrogen is required for normal mammary gland development, cumulative exposure to estrogen during a woman's lifetime is a high risk factor for breast malignancy. E2 also exerts protective effects against several diseases including cardiovascular diseases and neurological disorders.(1,2) The cytoprotective effect of E2 has primarily been attributed to its ability to induce key antioxidant defense enzymes as well as antioxidant properties that result from its phenolic structure.(3) The ability of cells to up-regulate their antioxidant enzymes is fundamental in the protection of cells and tissues from oxidative damage.(4) Such damage is potentially a major risk factor in the pathogenesis of various vascular diseases, cancers, and neurodegenerative disorders.
The ability of E2 to act as both a growth factor and a survival factor for breast cancer cells is not well understood, although anti-apoptotic function of E2 has been suggested as playing role.(5,6) Chemotherapy, a standard treatment for breast cancers, induces apoptosis. It has been reported that MCF-7 breast cancer cells acquire relative chemotherapeutic drug resistance against apoptosis in the presence of estrogen.(7) In this regard, it is of significance that E2 may provide a survival advantage in breast cancer cells. One aspect that has caused considerable interest is the effect of E2 on the expression of antioxidant enzymes involved in the clearance of reactive oxygen species (ROS). Studies suggest that E2 serves a protective role against oxidative stress in a variety of human tissues, including cardiovascular tissue,(8) neuronal tissues,(9) and liver.(10) Despite numerous studies on estrogens and breast cancer, there have been few reports on the effects of E2 on oxidative stress in breast cancer cells. Rao et al. suggested that E2-induced superoxide dismutase 1 may play a role in the survival of MCF-7 breast cancer cells.(11) Felty et al. demonstrated that estrogen-induced mitochondrial ROS modulate cell cycle progression in MCF-7 cells.(12) A low concentration of ROS can exert important physiological changes in cellular signaling and proliferation.(13) For example, growth factors (platelet-derived growth factor, epidermal growth factor and TGF-β), cytokines (IL-1 and TNF-α) and hormones (estrogens and insulin) are known to stimulate cell growth via the generation of ROS in many target cells.(14,15) This broad spectrum of responses to oxidants by proliferating cells suggests a new paradigm for oxidative stress.
It is well known that the cytotoxic effect of H2O2 is primarily caused by hydroxyl radicals (OH˙) generated from a series of reactions summarily called the Haber-Weiss/Fenton reaction. The OH˙ can trigger lipid peroxidation or the formation of lipid hydroperoxides, which are relatively long-lived ROS that are associated with cellular membranes.(16) Furthermore, once formed, lipid hydroperoxides mediate deleterious processes that lead to various cellular dysfunctions.(17) Therefore, to deactivate H2O2 and control the balance between the production and removal of oxygen radicals, cells contain a variety of antioxidant enzymes such as glutathione peroxidase (GPX), superoxide dismutase, catalase, and glutathione-S-transferase. Of these, GPX4 is a unique antioxidant enzyme in that cell membrane-bound phospholipid hydroperoxides are scavenged.(18) In addition, there is evidence suggesting that up-regulation of GPX4 expression and the subsequent increase in GPX4 activity may confer adaptive survival responses to oxidative stress. For example, GPX4-overexpressing RBL2H3 cells are reportedly resistant to necrotic and apoptotic cell death caused by various forms of oxidative stress.(6,18) Moreover, an in vivo model of this effect revealed that transgenic mice that overexpress GPX4 are protected against oxidative stress-induced apoptosis.(19) Therefore, in this study, we investigated the effect of E2 on the expression of the antioxidant enzyme, GPX4, as a mechanism of adaptation to oxidative stress in breast cancer cells. Our findings demonstrate that, in the presence of E2, estrogen receptor (ER)-expressing human MCF-7 cells induce relative resistance to oxidative stress. This process may stem from the finding that E2 increases basal ROS levels, which, in turn, stimulates the expression of GPX4.
METHODS
Reagents and cell culture
Hydrogen peroxide, 17β-estradiol, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrozolum bromide (MTT), ICI 182,780 (N-n-butyl-N-methyl-11[3,17β-dihydroxyoestra-1,3,5-trien-7α-yl] undecamide), 2',7'-dichlorofluorescin diacetate (DCF-DA), N-acetyl-L-cysteine (NAC), and antibody to β-actin were obtained from Sigma-Aldrich Co. (St. Louis, USA). LY294002 (2-([4-morpholinyl]-8-phenyl-4H-1-benzopyran-4-one) and PD98059 (2'-amino-3'-methoxyflavone) were purchased from Calbiochem (La Jolla, USA). U0126 was obtained from Promega (Madison, USA). Goat polyclonal antibody specific to GPX4 was obtained from Novus Biologicals (Littleton, USA). ERα antibody, rabbit anti-goat IgG-HRP antibody, and the enhanced chemiluminescence (ECL) system were purchased from Santa Cruz Biotechnology (Santa Cruz, USA). Trizol reagent and cell culture media were purchased from Life Technologies (Grand Island, USA). The human mammary adenocarcinoma cell line, MCF-7, was maintained in DMEM supplemented with 5% fetal bovine serum (FBS), 1 mM glutamine, 100 units of penicillin/mL and 100 µg of streptomycin/mL. Cells were grown to 70% confluence, after which the medium was changed to phenol red-free DMEM amended with 5% charcoal-stripped calf serum (CCS). The cells were then grown in a monolayer culture in this medium for 24 hr prior to treatment.
MTT assay
MCF-7 cells were seeded in 24-well plates in DMEM supplemented with 5% FBS and then grown to 70% confluence. Next, the medium was changed to phenol red-free DMEM amended with 5% CCS and the cells were then maintained in this medium for 24 hr. The cells were then pretreated with E2 (10 nM) for 24 hr, after which they were exposed to different doses (0-1,500 µM) of H2O2 for 24 hr. MTT (final 0.1 mg/mL) was then added to the culture medium, after which the cells were incubated for an additional 4 hr. Following removal of the medium, the formazan crystals formed as a result of the reduction of MTT by mitochondrial dehydrogenases present in the living cells were solubilized in 500 µL of DMSO and then measured spectrophotometrically at 555 nm. The results were expressed as a percentage based on the ratio of the absorbance of treated cells to that of the controls (100%).
Gene silencing with small interfering RNA (siRNA)
Control-scrambled-siRNA and ERα-specific siRNA were purchased from Dharmacon Research (Chicago, USA). The sequence of the ERα-siRNA was 5'-CCUCGGGCUGUGCUCUU-3'. Cells were seeded in six-well plates overnight, transfected with siRNA (50 nM) for 72 hr, and then treated with E2 (10 nM) for 24 hr. The effect of ERα gene silencing was then ascertained by Western blot analysis using anti-ERα and anti-GPX4.
Measurement of the intracellular reactive oxygen species
Intracellular ROS levels were measured using dichlorofluorescein diacetate (DCF-DA).(20) Briefly, MCF-7 cells were pretreated with vehicle (media) or NAC (10 mM) for 1 hr prior to incubation with E2 (10 nM) for the indicated times, after which the cells were loaded with 10 µM DCFHDA (Molecular Probes, Oregon, USA) for 20 min. Next, the cells were harvested and washed twice with 1X PBS to remove all of the excess DCF-DA that had not penetrated the cells. The relative fluorescence was then measured at excitation and emission wavelengths of 485 and 530 nm, respectively, using a multi-well fluorescence plate reader.
Western blot analysis for GPX4 expression
Cell lysates were prepared using a buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1% Nonidet P-40). Briefly, proteins (40 µg per lane) were separated on 10% Tris-glycine SDS-polyacrylamide gels and then electrophoretically transferred to Immuno-Blot PVDF membranes. The membranes were then incubated with a 1:500 dilution of goat anti-GPX4 polyclonal antibody for 1 hr at room temperature. Next, horseradish peroxidase-conjugated secondary antibody was applied at a dilution of 1:8,000. Anti-β-actin antibody was used as a loading control and the signal was visualized using an ECL detection kit.
RESULTS
E2 protects against H2O2-induced cell death and up-regulates GPX4 protein
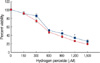
To confirm the cytoprotective effect of E2 against H2O2-induced oxidative stress, MCF-7 cells were exposed to different doses (0-1,500 µM) of H2O2 for 24 hr after being pre-incubated with E2. The viability of the cells was then measured by MTT assays. At higher concentrations than 300 µM H2O2, the viability of MCF-7 cells was significantly reduced (Figure 1). In comparisons, pretreatment with a 10 nM dose of E2 for 24 hr increased cell survival against subsequent exposure with H2O2. These results indicate that E2 can protect MCF-7 cells against subsequent lethal oxidative insults. We then examined whether increasing doses of E2 (from 0 to 100 nM for 24 hr) induce GPX4 expression levels in a dose-dependent manner. Western blot analysis data showed that E2-mediated GPX4 protein induction is dose-dependent, but it reaches plateau around 10 nM (Figure 2A). In time-course studies, a gradual increase of GPX4 protein expression levels by E2 was observed as time passes up to 24 hr (Figure 2B).
E2 modulates the GPX4 protein level by stimulating the generation of ROS
Several studies have shown that E2 stimulates the production of intracellular ROS, which is then involved in various signaling pathways.(21,22) Therefore, to determine if E2 can trigger the production of intracellular ROS and to explore the link between E2-induced ROS generation and GPX4 expression, we treated MCF-7 cells with E2 for various periods of time and then measured the intracellular ROS levels using the fluorophore, DCF-DA. As shown in Figure 3A, the intracellular ROS levels increased within 10 min of stimulation with E2 and then gradually declined until 60 min. However, pre-treatment with the antioxidant, 10 mM NAC, for 1 hr significantly reduced the E2-induced ROS levels in the cells. In consistent, pretreatment with NAC rapidly reduced the expression levels of GPX4 induced after E2 treatment for 1 or 24 hr (Figure 3B). Several growth factors and hormones are known to stimulate intracellular ROS production.(6) Thus, we investigated the effect of various growth factors and hormones on GPX4 protein levels. Our results showed that, in addition to E2, other ROS inducers including insulin, TGF-β and TNF-α increased intracellular GPX4 levels (Figure 3C). Taken together, the results of this study suggest that E2 regulates the levels of GPX4 protein in MCF-7 cells, at least partially, through a rapid and transient induction of ROS.
PI3K inhibition decreases E2-induced GPX4 up-regulation
Because the biological effects of estrogen are primarily mediated by estrogen receptors (ERs) through the transcriptional activation of genes containing estrogen response elements (EREs), we further examined the effect of ERα gene silencing on GPX4 expression following E2 treatment. MCF-7 cells were pretreated with 50 nM siRNA (control vs ERα) for 72 hr and then incubated with 10 nM E2 for 24 hr. As we expected, E2 treatment increased GPX4 protein expression in both no siRNA- and control-siRNA-treated groups (Figure 4A, 1 to 4th lane). However, E2 did not substantially induce GPX4 protein expression in ERα knockdown cells. These findings implicate that ERα-dependent pathways may be involved in an early induction of GPX4 expression within 24 hr of E2 treatment. In order to determine the role of the signaling pathways in the induction of GPX4, we used pharmacological inhibitors to block specific signaling pathways (ICI 182,780 for ER, PD98059 and U0126 for MEK1/2 and Ly294002 for PI3K). LY294002 effectively inhibited the expression of GPX4 that was induced by E2, while U0126 and PD98059 had no significant effect on the induction of GPX4 by E2 (Figure 4B). Taken together, these findings suggest that ERK1/2 is not involved in the expression of GPX4. Interestingly, up-regulation of GPX4 expression following E2 treatment was not clearly blocked by preincubation with the ER antagonist, ICI 182,780. Taken together, these findings indicate that signaling mediated by PI3K/Akt can play a significant role in GPX4 induction by E2 in MCF-7 cells.
DISCUSSION
The harmful effects of ROS are counterbalanced by cellular anti-oxidant enzymes such as superoxide dismutase, catalase, and GPX. Among these different enzymes, GPX4 is responsible for reducing lipid hydroperoxides and cholesterol ester hydroperoxides within cell membranes.(18) In particular, it has been reported that E2 has strong inhibitory activity toward lipid peroxidation.(23) In this study, we attempted to determine the molecular mechanism underlying the antioxidant effects of E2 by evaluating its up-regulation of GPX4 protein. Our findings demonstrated that treatment with E2 protects against H2O2-induced cell death, causes a low and transient accumulation of intracellular ROS levels, and induces the up-regulation of GPX4 expression in MCF-7 breast cancer cells. These results indicate that protection against H2O2-induced cell death in this cell line may be associated with the up-regulation of GPX4 protein, which is a part of the defense system of the organism under oxidative stress. The protective effects of E2 against oxidative stress have been widely reported.(24,25) It has also been demonstrated that overexpression of GPX4 protects cells against the apoptosis induced by hypoglycemia, arsenite, staurosporine, etoposide, cycloheximide, actinomycin D, 2-deoxyglucose, and UV irradiation,(4,26,27) as well as oxidative stress.(19) Furthermore, GPX4 protects against membrane damage via the reduction of cell membrane-bound phospholipid hydroperoxides.(18) In particular, it has been reported that E2 has strong inhibitory activity toward lipid peroxidation.(23) Taken together, these findings imply that the cytoprotective role of E2 is linked to its ability to induce the intracellular antioxidant protein, GPX4.
It is widely accepted that low levels of H2O2 and the related ROS play essential roles in cell signal transduction and can induce an adaptive response.(28,29) The results of the present study revealed that E2 induced a rapid and transient increase in the level of intracellular ROS that occurred within 10 min of treatment, peaked after 15 min, and then declined after 1 hr. The induction of intracellular ROS production in response to treatment of MCF-7 cells with E2 in this study is consistent with the results of previously conducted studies.(30) In addition, we did not detect any significant cytotoxicity in MCF-7 cells that had been treated with E2 for 24 hr, which indicates that a mild increase in intracellular ROS production may enable signal transduction, but may not be sufficient to induce oxidative stress. The roles that ROS play in the modulation of signal transduction have recently received increased attention. For example, several studies have shown that stimulation of diverse receptor systems including those of TNF-α, TGF-β and insulin induces ROS, which may serve as requisite secondary messengers.(14,15) Interestingly, these molecules also induced an increase in the GPX4 protein level, whereas KGF had no effect on the GPX4 level. Although all of the molecules evaluated in this study, with the exception of KGF, were already known to stimulate the production of intracellular ROS, our results are the first to suggest that intracellular ROS are involved in the regulation of GPX4 by E2. Furthermore, the free radical scavenger, NAC, rapidly inhibited the E2-induced up-regulation of GPX4 protein, which indicates that ROS play a critical role in the regulation of GPX4. Therefore, it would be rational to assume that a low level of ROS induced by E2 serves as a signal that warns cells of the oxidative status, thereby triggering up-regulation of GPX4 expression.
Many, though not all, of the physiological effects of E2 are mediated through transcriptional activation of genes containing EREs. Therefore, to determine if GPX4 expression is regulated by ERα-mediated transcriptional activity, MCF-7 cells were transfected with ERα-siRNAs. We found that E2-induced GPX4 induction decreased in ERα knockdown cells. However, we also obtained somewhat controversial data that pretreatment with the ER antagonist, ICI 182,780, does not efficiently block E2-induced GPX4 expression. Studies that examine the interaction of ER on GPX4 promoter are necessary to better understand this relationship. Among the local E2 effects, there is much evidence that suggests mechanisms independent of ER activation. For example, it has been shown that E2-stimulated ROS production is not correlated with the activity of ER in MCF-7 cells.(21) Furthermore, Lee et al.(30) suggested that ER-independent antioxidant effects contribute to the protective role of E2 against hypoxia-induced hepatocyte injury.
Interestingly, similar to the effects of the antioxidant, NAC, GPX4 expression by E2 was strongly blocked in response to pre-incubation with the PI3K inhibitor. This finding suggests that PI3K/Akt signaling plays a role in the regulation of GPX4. Furthermore, previous studies have shown that ROS accumulation induced by TNFα,(14) PDGF,(31) or VEGF(32) was suppressed when PI3K activity/activation was blocked by pharmacological or transfectional means. However, some studies have reported the ability of ROS to activate PI3K in various cell types.(33-35) Although we did not define the difference, a positive feedback mechanism appears to be involved in the mutual interaction that occurs between ROS and PI3K, and this mechanism appears to act in a cell type-specific manner. Therefore, it should be noted that GPX4 may be a point of convergence of the Akt and ROS signaling pathways. However, it is unclear if the ROS and Akt signals cross-communicate with each other or operate on alternative pathways. Overall, we report that the effects of E2 on GPX4 expression are, at least partially, regulated by PI3K/Akt signaling and/or low levels of ROS. Although there is limited information regarding the signaling mechanisms that modulate GPX4 levels, our findings offer important information regarding the underlying mechanism by which E2 protects cells against oxidative damage in breast cancer cells.
CONCLUSION
Our studies have led us to hypothesize that the rapid and profound E2-mediated induction of GPX4 protein can be an important step in the adaptation of MCF-7 cells to oxidative status. In breast cancer cells, Estrogen deprivation may facilitate the effects of oxidative stress-based therapies through reduction of cellular antioxidative capacity and offer therapeutic benefits.



 XML Download
XML Download