

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Introduction
Sepsis, the leading cause of death among critically ill patients, is a syndrome characterized by initial exaggerated systemic inflammatory response to infection or trauma followed by relatively prompt immune dysfunction. In acutely ill patients, a robust inflammatory response to severe sepsis is typically linked with multiple organ dysfunction syndrome (MODS), whereas septic shock is in addition associated with hypotension that does not respond to fluid resuscitation and vasopressors [12]. The mortality rates are from 25 to 30% for severe sepsis and up to 30-40% in shock [34]. Importantly, while early diagnosis and antibiotic therapy improved survival, post-sepsis complications have devastating impact. In particular, cognitive impairment, physical disabilities and frailty link to susceptibility to injury and infections, as well as poor recovery are associated with high mortality among sepsis survivors [5].
Organ dysfunction from sepsis is traditionally attributable to the effects of inflammatory mediators and tissue hypoxia and cell damage [678]. Organ injury is especially detrimental in lung and kidney, where regenerative capacity is limited. Surprisingly, several studies have found a substantial discordance between histological analysis and the degree of organ failure in patients who died from sepsis [91011]. Little or no apoptosis or necrosis was found and in many cases tissue oxygen delivery was preserved [9]. Notably, most clinical trials revealed little or no effects of the anti-inflammatory therapy [1213]. Low efficacy of anti-inflammatory drugs raises many questions about duration of tested therapies, pre-existing conditions and apparently multifactorial causes of sepsis. However, it is suggested that despite of oxygen bioavailability sepsis has a profound impact on cellular metabolism along with impairment of mitochondrial function [1415]. In particular, a reduced cellular respiration, also termed “cell hibernation” or “cytopathic hypoxia”, may be initially linked to cells survival, but afterward can trigger to multi-organ failure in sepsis [1617]. The concept was developed by early studies that found abnormal swollen mitochondria in animal model of sepsis [18]. Subsequently, it has become apparent that sepsis is characterized by a reduced function of the mitochondrial respiratory chain and accompanied by free radical generation and bioenergetic reprograming toward glycolytic metabolism [1920].
Sepsis has a tremendous impact on the immune system by affecting leukocytes pro-inflammatory activity, microbial eradication, viability and proliferation [2122]. In the initial phase of infection or trauma, immune response encounters hyperinflammation. This event is associated by excessive accumulation of inflammatory cytokines and damage associated molecular pattern proteins (DAMPs) in tissue fluids and plasma [2324]. The cytokine storm is also described as systemic inflammatory response syndrome (SIRS), that characterized an excessive release of inflammatory cytokines, including interleukin (IL) 1 and 17 and tumor necrosis factor alpha (TNF-α) [25]. However, initial leukocyte activation is leading to immune dysfunction and development of immunosuppression [26]. Critically ill patients often progress to immunosuppression within a few hours of sepsis diagnosis followed by increased susceptibility to lung infection persisting for several days or weeks. In addition to high risk of nosocomial infections, septic patients developing chronic critical illness for greater than 14 days progress to persistent inflammation, immunosuppression and catabolism syndrome (PICS). These conditions are associated by following ~20-40% mortality rate by the 2-year mark [2728]. It is important to note that over 2.5 million survivors of severe sepsis, septic shock, and PICS are at risk of lung infection and death [28].
Recent advances in clinical and experimental sepsis provided a wide range of discoveries related to immunometabolism and bioenergetics. However, despite economic and social burden of sepsis, therapeutic interventions are limited, mostly to early administration of antibiotics and fluids resuscitation. An effective treatment for critically ill patients or to improve quality of life among sepsis survivors is crucially needed. In this review, we highlight metabolic alterations in immune and organ tissue homeostasis, and indicate emerging therapeutic opportunities.
Mitochondrial function and alignment with glucose oxidative metabolism
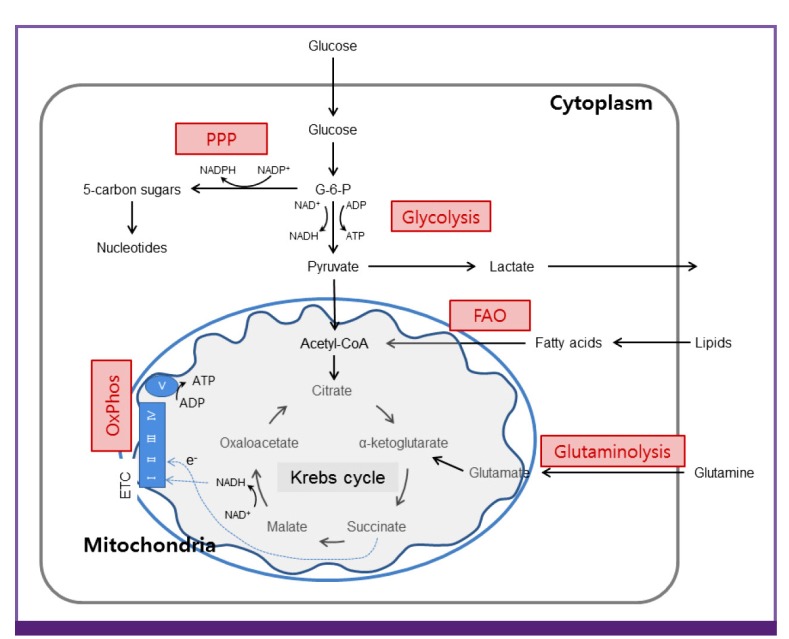
Mitochondrial bioenergetics consist oxidative phosphorylation (OxPhos) integrated with the glycolytic pathway of the Krebs cycle (Fig. 1). Glucose enters the cellular milieu through the glucose transporter 1 (Glut1) followed by conversion to pyruvate that is mediated by series of enzymatic steps, including glucose phosphorylation to glucose-6-phosphate (G-6-P) followed by conversion to pyruvate, reducing NAD+ to NADH and generating two ATP molecules. Following its transportation into the mitochondria through converted into acetyl-CoA, pyruvate is further oxidized to CO2 via the Krebs cycle to generate NADH that is oxidized via the OxPhos. In particular, mitochondrial electron transfer chain (ETC) complexes are essential for generation of mitochondrial membrane potential and proton gradient that is further utilized for production of ATP at the complex V (ATP synthase). In addition to the breakdown of glucose via glycolysis, cells have the ability to metabolize other substrates, such as lipids and glutamine, which feed into the Krebs cycle and drive OxPhos. Fatty acid β-oxidation and glutaminolysis replenish the Krebs cycle intermediates acetyl-CoA and α-ketoglutarate, respectively, thereby fueling oxidative phosphorylation. Of note, during inflammation and/or reduced oxygen, ATP production is derived by breakdown of glucose due to glycolysis and pyruvate being routed toward lactate instead acetyl-CoA. In sepsis, this situation is associated with increased amounts of tissue localized and in systemic lactate, though impaired lactate clearance is also a contributing factor [2930].
Figure 1
Integration of metabolic pathways
Glucose is metabolized to pyruvate through the glycolysis. Pyruvate (and fatty acids) enters the mitochondria where they are converted to acetyl-CoA. This enters the Krebs cycle that donates electrons to electron transport chain. Through OxPhos, electrons are sequentially transferred to generate a H+ gradient across the inner mitochondrial membrane, which drives the synthesis ATP. In addition to the glycolysis, cells have the ability to metabolize alternative substrates, such as lipids and glutamine. FAO and glutaminolysis replenish the Krebs cycle intermediates acetyl-CoA and α-ketoglutarate, respectively, thereby fueling OxPhos. PPP generates riboses for nucleotides synthesis.
PPP, pentose phosphate pathway; OxPhos, oxidative phosphorylation; FAO, fatty acid β-oxidation; ADP, adenosine diphosphate; ATP, adenosine triphosphate; ETC, electron transport chain.
Mitochondrial dysfunction correlates with sepsis-related multi-organ failure
Along with bioenergetics, mitochondria are involved in several crucial functions that include program cell death pathway, calcium flux and redox signaling [31323334]. The exact reason for mitochondrial dysfunction during sepsis is not well understood. However, inflammatory molecules such as nitric oxide (NO), carbon monoxide, and reactive oxygen/nitrogen species directly impair several components of the mitochondrial ETC complexes and mitochondrial respiration [163536]. Additionally, lower metabolic rates in sepsis have been associated with decreased the amounts of mitochondrial DNA and expression of major components of ETC complexes [37]. This is significant issue because mitochondrial DNA code nearly eighty percent of mitochondrial protein. Besides decreased amounts of major components in mitochondrial respiratory chain complexes and ATP synthase, recent studies have shown diminished pyruvate dehydrogenase expression in sepsis and ARDS [383940]. It is important to note that pre-existing factors contribute to the severity of sepsis, including cigarette smoking, environmental exposure to toxins, metabolic syndrome of diabetes and obesity and aging [414243].
Clinical analysis of sepsis by Dr. Mervyn Singer laboratory has shown that the extent of mitochondrial impairment in lungs was correlated with mortality rate. In particular, sepsis-associated mortality is significant in patients that develop acute respiratory distress syndrome (ARDS) [4445]. Patients who died from severe sepsis had decreased muscle ATP content while higher levels of ATP were seen in survivors [45]. Organ dysfunction and clinical illness were accompanied by decreases in metabolic rate and mitochondrial mass [37]. However, recovery of metabolic activity and organ function is possible, and were strongly regulated by expression of markers of mitochondrial biogenesis such as PRARgamma-coactivator-1a (PGC-1a), nuclear respiratory factors 1 and 2 (NRF-1 and -2), and via repression of the biogenesis suppressor nuclear receptor interacting protein-140 (RIP140) [37464748]. Moreover, most recent pre-clinical studies, and our results, indicated that not only preservation, but also mitochondrial biogenesis is an essential step in reestablishing immune or tissue organ homeostasis during recovery from sepsis [495051].
Metabolic control of immune cell homeostasis and pro-inflammatory activation
Severe infections, trauma and hemorrhage are initially associated with hyperinflammatory state, which frequently leads to immunoparalysis. This situation is implicated in nosocomial infections (high risk of hospital-related infections), and community acquired pneumonia [2652]. Recently, it has become apparent that these events are correlated with specific metabolic and bioenergetic alterations in immune cells [5354]. Many immune cells lose their bioenergetic plasticity due to glycolytic and anabolic metabolism. Leukocytes from patients with severe sepsis show deficient cellular metabolism that were associated with a defective response to secondary stimulation. Notably, recent studies suggest that both glycolysis and OxPhos are impaired in monocytes of post-septic immunosuppressed patients [5556].
1. Neutrophils
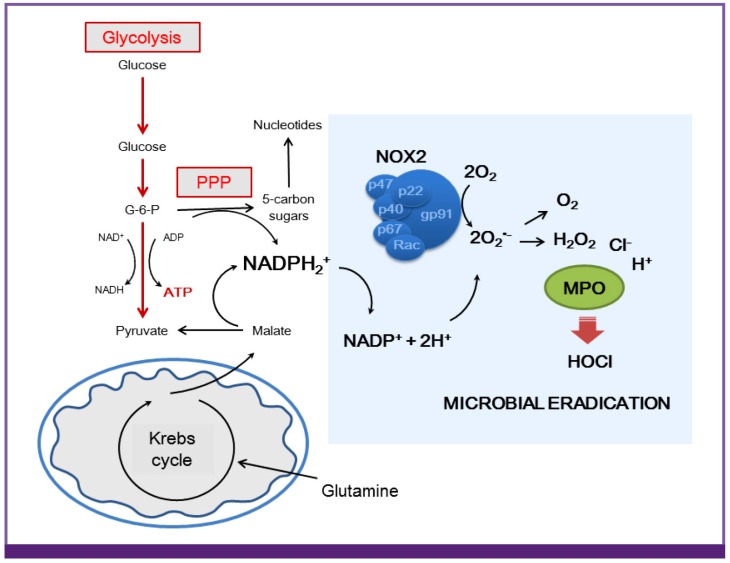
Neutrophils are most abundant leukocytes in circulation. Along with macrophages, they are a first line of innate immune response during microbial infections in traumatic injury [57]. Mitochondria were relatively recently described in neutrophils. They have relatively low density and negligible oxygen consumption rate [58]. Indeed, neutrophil pro-inflammatory action is predominantly supported via an extensive glucose utilization [59]. Activated neutrophils also synthesize NADPH, an essential cofactor for the NADPH oxidase 2 (NOX2) function specifically directed to generation of the superoxide and subsequently hydrogen peroxide (H2O2) (Fig. 2) [6061]. Besides glycolysis, neutrophil bioenergetics is also partially supported by the pentose phosphate pathway (PPP) of glycolysis and glutaminolysis. Activation of Hypoxia-inducible factor-1α (HIF-1α) and the mammalian target of rapamycin (mTOR) are major metabolic signaling components that control neutrophil glycolytic phenotype [6263]. For example, HIF-1α activates the Glut 1 and also control the expression of a number of antimicrobial factors in neutrophils, including granule proteases, antimicrobial peptides, NO, and TNF-α [6465]. When innate immune cells are deficient in myeloid-specific HIF-1α, mice are not protected against Staphylococcus aureus sepsis, indicating that this HIF-1α pathway and glycolytic flux are integral for septic immune responses [2466].
Figure 2
Neutrophils anti-microbial action is linked to bioenergetics
NADPH, which is required for production of ROS, is generated in PPP branch of glycolysis or from the oxidation of glutamine derived malate to pyruvate. In this manner, the biochemical pathways of glycolysis and glutaminolysis provide the microbicidal activity to activated neutrophil.
PPP, pentose phosphate pathway; ADP, adenosine diphosphate; ATP, adenosine triphosphate; ROS, reactive oxygen species; MPO myeloperoxidase; NOX2, NADPH oxidase 2; HOCI, hypochlorous acid.
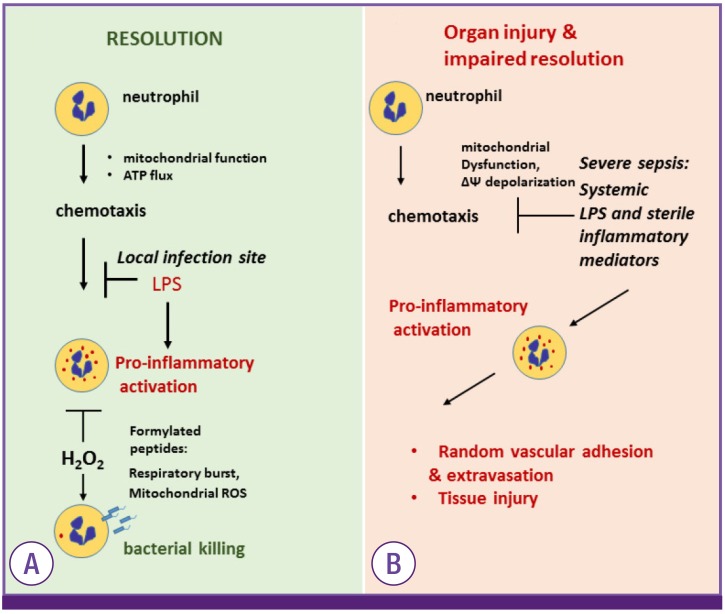
Interestingly, while mitochondria have little or negligible impacts to neutrophil bioenergetics, recent studies indicate that proximal localization of mitochondria in the leading edge is crucial for optimal chemotaxis [676869]. In particular, dissipation of mitochondrial membrane potential, a situation found upon exposure to bacterial product LPS, nearly completely diminished neutrophils chemotaxis [5070]. This is relevant issue because host-generated inflammatory mediators and bacterial products are present in circulation of individuals with established sepsis. These mediators effectively prevent neutrophil chemotactic response and cause random adhesion to epithelium, and thereby cause vascular injury (Fig. 3) [71]. Previous studies have indicated that mitochondrial membrane depolarization also reduced neutrophil respiratory burst. Although this issue is directly linked to inability to kill bacteria, mechanistic insights remain to be determined [5172]. It is important to note, ROS produced by respiratory burst, likely mitochondrial source, or simple exposure to extracellular hydrogen peroxide bolus have a significant impact on neutrophil pro-inflammatory function. Apparently, H2O2 is indispensable for an effective microbial eradication, but it is also preventing and/or promoting neutrophil transition from pro-inflammatory to anti-inflammatory phenotype that linked to bacterial killing (Fig. 3) [7374]. Collectively, these new findings clearly indicate that mitochondria play a crucial role in neutrophil biology.
Figure 3
Mitochondria plays a crucial role in regulating neutrophil function.
(A) Mitochondrial function in the leading edge supports neutrophil chemotaxis. In infection site, LPS-TLR4 engagement indices mitochondrial depolarization and subsequent inhibition of neutrophil motility and promote pro-inflammatory action. Pro-bactericidal effects are activated by NADPH oxidase activation. Notably, H2O2 is important to bacterial killing and promote neutrophil transition from pro-inflammatory to anti-inflammatory phenotype. (B) Severe sepsis and shock is characterized by accumulation of bacterial products and DAMPs in circulation. LPS inhibits neutrophil chemotaxis and promote pro-inflammatory activation flowed by random adhesion to endothelial cells. Neutrophils extravasation and activation is implicated in tissue injury.
ATP, adenosine triphosphate; TLR, Toll-like receptor; LPS, lipopolysaccharide; NADPH, nicotinamide adenine dinucleotide phosphate; DAMPs, damage associated molecular pattern proteins.
2. Macrophages and dendritic cell
In the presence of pathogens or trauma and ischemia, macrophages rapidly switch from a resting state to a highly active pro-inflammatory state. This is associated with increased cytokines, chemokines and production of other host defense factors that enhanced phagocytosis, and later on antigen presentation. TLR4-LPS engagement is associated with acquisition of pro-inflammatory phenotype, which often called classical (M1) macrophages [75]. M1 macrophages have a high microbicidal activity and are characterized by production of pro-inflammatory cytokines and ROS. Similar to neutrophils, HIF-1α is a primary regulator of glycolytic metabolism in M1-polarized macrophages. Another important regulator of macrophage activation is carbohydrate kinase-like protein (CARKL) which is typically reduced in M1 [76]. In turn, CARKL expression promotes exit toward M2 phenotype, in which the induction of pro-inflammatory cytokines is greatly diminished [75]. This phenotype can be directly elicited by stimulation macrophages with IL-4 and IL-13. M2-polarized macrophages also participate in host defense, including neutralization of parasites, infections and stimulate tissue repair by production of anti-inflammatory cytokines and phagocytosis of dying cells [7577]. Indeed, poor transition from M1 to M2 phenotype has detrimental impact on recovery from organ injury. For example, it has been shown that deficiency of AMP-activated protein kinase (AMPK) is directly implicated in impaired M1-to-M2 transition and disrupted clearance of apoptotic cells during the resolution phase [78798081].
M1 and M2 activation are characterized by a distinct metabolic profile that differs from resting macrophages (M0) [82]. In particular, M1 macrophages have high rates of glucose and glutamine uptake and lactic acid production with little or no flux through OxPhos [8384]. In activated macrophages, OxPhos is inactivated following the inducible form of nitric oxide synthase (iNOS) dependent NO production; NO competes with oxygen to inhibit the terminal electron acceptor (complex IV) of mitochondrial electron transport chain [8586]. Our recent studies indicate that preservation of major components of mitochondrial complexes is possible in a polymicrobial intra-abdominal model of sepsis [5087]. For example, metformin is major metabolic AMPK activator that promoted mitochondrial biogenesis and thus, decreased the severity of endotoxin induced acute lung injury [50]. Notably, MAPK kinase 3 inhibition effectively preserve mitochondrial function on lung of mice subjected to endotoxin injury [88]. These finding suggest that preservation/restoration of mitochondrial function was essential for immune recovery from sepsis. Interestingly, an increment in mitochondrial ROS production is required for normal macrophage bactericidal activity [89]. In regards to the metabolic phenotype, M2 macrophages had higher rates of fatty-acid oxidation (FAO), mitochondrial biogenesis [56]. Compared to HIF-1α dependent glycolytic metabolism of M1 macrophages, equation of M2 phenotype is associated with FAO and oxidative metabolism. These are mediated by at least in part by activation of STAT6 [56] that promotes expression of genes involved in FAO and OxPhos due to mitochondrial biogenesis mediated by peroxisome-proliferator-activated receptor-γ co-activator-1β (PGC-1β) [56]. Moreover, expression of peroxisome proliferator-activated receptor (PPAR) is increased in M2 macrophages, and this supports the view that PPAR are important transcription factor that drive transition to the M2 phenotype [90].
Similarly to macrophages, dendritic cells (DC) oxidize glucose in the mitochondria through OxPhos and in response to TLR agonists. They also activate the metabolic switch for glycolytic metabolism during pro-inflammatory activation [84]. An initial increase in glycolysis occurs within minutes of DC activation. Over the course of 18 hours, activated DC sustains elevated glycolysis along with loss of OxPhos. This metabolic shift appeared to have a substantial impact in regulating DC-induced T cell responses [91].
Although above paragraph summarized major characteristics of M1 and M2 phenotypes, it is important to note that several intermediate or phenotypic deviations are likely implicated in pathophysiological conditions. For example, M1-deriven pro-inflammatory response is implicated in inflammatory organ injury, at least in a murine model of sepsis. In turn, a subsequent transition to M2 may contribute to immunosuppression. This raises a question whether these adverse effects are mediated by bioenergetic adaptations. However, a plausible explanation may be derived from two recent studies that suggest dysfunction of both oxidative phosphorylation and glycolysis occurred in immunosuppressed monocytes [5556]. Another intriguing question is how macrophage M2 polarization accelerates resolution from organ injury, but it may also promote adverse fibrogenic remodeling. Indeed, these areas of research have recently revealed that several intermediate phenotypes of macrophages are possible [9293]. However, current models provide unclear explanation how bioenergetics profile of immune cells support normal resolving conditions, but also can promote disease progression. This is likely linked due to local tissue environment. Further studies are needed to delineate if bioenergetic profiles of M2 macrophages is different in M2 participating in essential normal wound healing vs. M2 associated with pathological fibrosis. Similar concerns are related to participation of M2 phenotype in resolution of inflammatory conditions, though is also suggested to elicit the immunosuppressive effects.
3. T cells
A hallmark of a successful immune response is the generation of memory T cells which are antigen-specific and can be rapidly activated to respond quickly to pathogen [94]. Indeed, production of regulatory T cell (Treg) contributes to T-cell inactivation and increase severity and mortality in experimental sepsis and post sepsis complications [9596]. T cells differ from innate cells in two important functions. First, they proliferate extensively and rapidly upon antigen specific activation. Secondly, after completion of the immune response, a subset of lymphocytes generates a long-lived, antigen-specific memory cells that mediate protection against reinfection [6194]. As expected, the metabolic profile of these each distinct subset of lymphocytes are different depend on their bioenergetics demands. In particular, naïve T cell relies on glucose oxidation through OxPhos [979899100]. This bioenergetic profile is significantly altered when naïve T cells are stimulated through antigen or cytokine receptor-dependent mechanism. Stimulated cell undergoes rapid growth, proliferation, and acquisition of specialized effector functions. These events are supported by enhanced glycolysis and glutaminolysis [101]. For example, CD28 stimulation is linked to PI3K/Akt –dependent increases in the abundance of Glut1 on T cell membrane surface [101]. This not only enhances the uptake of glucose by activated T cells but also promotes a switch from OxPhos to glycolytic metabolism via mammalian target of rapamycin (mTOR) [94101]. CD3, specific T cell receptor, also stimulate proliferation and strong dependence on extracellular glutamine [102]. In this study, Wang et al. observed that stimulation of resting T cells increases glycolytic flux through PPP, and glutaminolysis, while suppressing oxidation of pyruvate and fatty acids. Subsequent study revealed that although HIF-1α and Myc are rapidly induced upon T cell activation, Myc is primarily required for glycolysis, glutaminolysis, and T cell proliferation [103]. It is well-defined that following activation and proliferation, T cells differentiate into T helper (Th) 1, Th2, Th17, or Treg subsets. Activated T helper cells use glycolysis to support their effector functions [104], whereas Treg cell predominantly use OxPhos and mitochondrial FAO for development and survival [105]. Memory T cells share many of the same characteristics of naïve cell; they are long-lived, relatively inert cells with limited biosynthetic demands. Although both naïve and memory T cells are dependable on oxidative metabolism, memory T cells are metabolically unique because memory T cells are quiescent but positioned to respond rapidly. Memory T cells predominantly use FAO to generate acetyl-CoA to fuel OxPhos [106107].
Metabolic pathways can influence not only activation of T cells, but also the development of various T helper subsets. The transitions of T cells from naïve to activation and back to memory formation are highly dependent on and regulated by cellular metabolism in response to environmental signals.
Extension of metabolic profile to diagnose outcome of sepsis
Lactate is an established marker of bioenergetic alterations in predictor of mortality in sepsis. However, recent studies indicate that patents with prior use of metformin have shown nearly 30% increase survival despite high level of lactate [108]. Notably, while lactate is among well-established predictors of sepsis, recent clinical trials have shown that decrease in lactate production had no effects on sepsis survival [109110]. Clinical studies also suggest that lactate clearance may not be used as a surrogate marker of microcirculatory blood flow [111]. Several groups have performed broader metabolic profiling in sepsis to test whether incorporation of multiple metabolites could serve as prognostic indicators in sepsis, including identification of citrate, malate, glycerol, carnitine, sucrose, mannose, methionine, arginine and other metabolites [112113114]. Further metabolomic studies provide potentially valuable diagnostic markers to more accurately predict outcome in septic patients [113115]. In particular, analysis on plasma samples for FAO, gluconeogenesis, and the Krebs cycle between revealed a defect in FAO in non-survivors [113]. Apart from the direct role of metabolites in insuring the cellular energy resources, metabolites have an underestimated their role in signaling pathways and regulation of gene expression [115]. The interaction between metabolic pathways and the epigenetic profile of the cells plays an important role in the inflammatory phenotypes of immune cells during sepsis [115]. One of the most extensively described is the deacetylase enzymes SIRT1 in sepsis. TLR4 signaling results in SIRT1 binding to NF-κB to decrease NF-κB dependent transcription of pro-inflammatory cytokine [116].
Given the mitochondrial bioenergetics dysfunction and mortality rate in sepsis [1617], main metabolic switch AMPK, mTOR, HIF-1α and other regulator of glycolysis are among important therapeutic targets. For example, recovery of AMPK activity reduced the severity of sepsis and lung injury. AMPK also improved bacterial eradication in mouse model of peritonitis or intra-abdominal bacterial sepsis [70117118]. In part, AMPK activation reduced cytokine production, inhibited DAMPs release such as HMGB1 release [119]. Of note, AMPK also reduced immunosuppression through diminishment of HIF-1α and preservation of sensitivity of macrophages secondary challenge with LPS, thus reduced their immunosuppressive phenotype [50117120121]. In turn, Cheng et al. have shown that HIF-1α is important for preservation immune cell function when oxygen level is limited [66]. Notably, AMPK activator AICAR used in this study has substantial off-site effects related to adverse impact on immune chemotaxis, similar to AMP, GMP and other nucleotides [66]. Moreover, recent study uncovered that specific mechanism(s) are responsible for inactivation of AMPK activity during sepsis and in immunosuppressed monocytes. Therefore, typical AMPK activators may provide optimal effects as recently observed in mouse fungal infection model [55]. Notably, targeting glycolytic pathway, including pyruvate kinase M2 (PKM2) effectively reduced the severity of sepsis [122123]. Although this is exciting and promising area of research, it is not clear whether prior recovery of mitochondrial function is required for benefits mediated by reduced glycolytic flux toward restoration of immune homeostasis.
Conclusions
Severe sepsis and septic shock induce profound metabolic alterations and loss of bioenergetic plasticity in immune system. The pro-inflammatory phase and subsequent development of immunosuppression appear to have deficiency in mitochondrial bioenergetics of immune and parenchymal cells. Thus, pharmacological interventions that improve mitochondrial biogenesis and quality control are likely relevant for sepsis and other inflammatory conditions related to loss of bioenergetic and metabolic plasticity.
 XML Download
XML Download