

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Introduction
No one can deny that there is a cause for every disease in humans. All infectious diseases have been linked to causative pathogens, but a precise immunopathogenesis of most infectious diseases remains illusive. Most of the diseases have characteristic histopathological findings, with identical sets of responsive immune cells within each individual. It is believed that the main roles of the immune/repair system of the host are to prevent tissue cell injuries and to repair tissue damage from various insults resulting from the infection. Because circulating immune cells and proteins, such as immunoglobulins and complements, are only effectors for control of tissue cell injuries caused by various pathogens, it could be inferred that immune cells and antibodies found in pathologic lesions may control the toxic substances and function similarly in response to insults from various pathogens. Communication between immune cells, through major histocompatibility complexes (MHCs) and cytokine networks, is indispensable for efficient and complete immune reaction against any infectious insults.
The human body is considered a microcosm and maintains homeostasis for a healthy state via a responsive controlling network. The author has previously proposed that this network control is performed by the protein homeostasis system (PHS), and the immune system is one component of the PHS in organisms. The author has also briefly discussed the immunopathogenesis mechanisms of several infectious diseases including Mycoplasma pneumoniae pneumonia, influenza pneumonia, mumps, and Kawasaki disease (KD) through the PHS hypothesis [1, 2, 3, 4, 5]. In this paper, the author proposes a unified model of immunopathogenesis for nearly all infectious diseases through the PHS hypothesis, based on contributions from various biological and medical fields and personal clinical observations of various childhood diseases.
Go to : 
Cells, their receptors, and binding proteins
Multicellular organisms consist of various organs, and organ tissues consist of specific cells that have their own profiles (and cell fates), including different kinds and timings of gene expression (protein production) and presence or absence of specific cell receptors. All of the phenomena occurring in organisms, including embryonic development, biochemical, physiological and pathological phenomena, are regulated through proteins acting in a specific order. One of the most basic concepts for biology at the cellular level is the "receptor-signal transduction system." Substances (mainly proteins, as ligands that have specific affinities) bind to receptors on and in the cells, and induce the production of new proteins that may be needed for cells or organisms. Various sizes of protein derivatives made in vivo, including monoamines, peptide hormones, cytokines and larger proteins, bind cell membrane receptors and ultimately lead to the expression of new proteins. These newly expressed proteins may control intracellular events and/or they could bind receptors on other cells, if extracellularly released, and lead to a new cascade of protein expression elsewhere in an organism. Because each cell in mammals is protected and separated by a cell membrane, this basic biological activity is indispensable for communication between cells. On the other hand, there must be a control system that prevents endless protein production and that maintains protein homeostasis in cells and in the whole organism. There are many epigenetic factors that influence the homeostasis of a cell and a whole organism. Among them, micro RNAs and DNA methylation are important for gene activation or gene silencing, and regulate protein production. However, these two epigenetic mechanisms are also controlled by various protein enzymes, including RNA polymerases and DNA methytransferases [6, 7]. These findings reveal the complexity and importance of cross-talk between proteins (protein homeostasis) at the cellular level. Again, protein homeostasis should be maintained not only at the cellular level but also systemically for maintenance of the healthy state of an organism (Fig. 1).
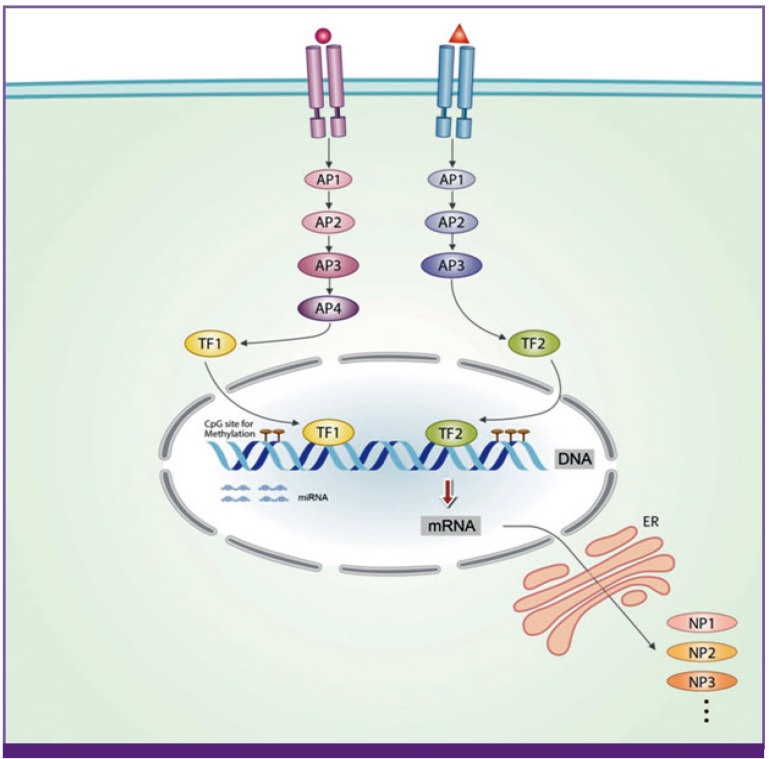 | Figure 1The receptor-signal transduction system at cell level, as a basic principle of all biologic phenomena.Each organ-specific cell is protected by a cell membrane and has its specific receptors. When the substances, mainly proteins, bind to these receptors, the substance-receptor complex transmits the signal to the nucleus through activation of a series of adaptor proteins and transcriptional factors and produces new proteins. The binding substances include various sizes protein derivatives such as monoamines, peptide hormones, cytokines, exogenous toxins from pathogens such as pathogen associated molecular patterns (PAMPs), non-protein biochemical substances such as vitamins and essential fatty acids, and chemical substances such as drugs. This basic structure of the cells may be essential to protect the cells of the host, and communicate across cells through proteins (cytokines) including self-identifying proteins such as major histocompatibility complexes (MHCs). Epigenetic processes such as gene methylation and microRNAs may affect on production of such proteins, but these processes are also regulated by other enzymatic proteins within the cells, suggesting a exist of protein homeostasis system (PHS) in cells. The protein homeostasis should be maintained at systemic level for combating against pathogens and maintaining of healthy state of the organism. AP, adaptor protein; ER, endoplasmic reticulum; TF, transcriptional factor; NP, new protein; miRNA, microRNA; mRNA, messenger RNA.
|
Natural invaders, such as infectious pathogens and animal venoms, are exogenous proteins that are toxic to host cells and consist of protein complexes of variable size expressing different levels of virulence. Endogenous proteins that are released from activated or destroyed cells, and those produced in an immune response, including proinflammatory cytokines and proteolytic enzymes from immune cells such as macrophage or granulocytes, could also be toxic to the host cells. The "cytokine storm" that occurs in various infections, such as those of the Ebola virus or influenza virus may be an example of extreme cases [8]. Proteins that are harmful to the host cells can be designated as pathogenic proteins (PPs), and can also be classified as exogenous and endogenous PPs [5]. There are several known diseases by which the damage to cells of one organ affects the cells of the same/other organs. The historical example is sympathetic ophthalmia. After traumatic injury to one eyeball, inflammation can occur within both ocular tissues [9]. Thus, substances which induce inflammation of normal eye tissues may originate from injured eyeball tissue cells, and they may reach target cells in the opposite eye via systemic circulation. Rhabdomyolysis is another example, and this acute muscle cell injury is associated with various insults, including trauma and infections such as measles and influenza [10]. Rhadomyolysis can lead to complications such as acute kidney injury, disseminated intravascular coagulation and arrhythmia, which are all due to systemically circulating substances derived from the injured muscle cells, including myoglobin, potassium and other toxic materials. Keloids and hypertrophic scars result from abnormal fibrous wound healing after skin injuries. However, the etiologic origin of this skin disorder may be materials derived from injured skin cells, as well as maladjustments from responding immune cells [11]. Additionally, activated neutrophils secrete proteins, including myeloperoxidases and matrix metalloproteinase-9, and activated eosinophils also secrete proteins, including major basic protein and eosinophil cationic protein. These proteins can be toxic and can induce inflammation in various tissues [12, 13].
There are over a hundred thousand kinds of proteins in human serum, from monoamines to larger lipoproteins, but a majority of these are small proteins i.e., peptides [14]. Major neurotransmitters such as dopamine, norepinephrine and serotonin of the central nervous system (CNS) are monoamines and may have important roles in CNS embryogenesis and the homeostasis of CNS cells [15]. Neuropeptides such as thyrotropin-releasing hormone (tripeptide), enkephalin (pentapeptide) and oxytocin/vasopressin (nonapeptides) in the CNS or endocrine system, and some peptide substances such as angiotensin I (decapeptide) and angiotensin II (octapeptide) in the renin-angiotensin-aldosterone system, are composed of 3-10 residue long peptides, and these peptides maintain hormonal homeostasis of the host through the receptor-signal transduction system [16, 17]. Therefore, it is a reasonable assumption that peptides in blood are composed not only of pieces of larger proteins but also of those having biological activities that remain to be defined in future studies. In addition, there is increasing evidence that some peptides directly affect host cells through the receptor-signal transduction system. For example, among natural toxins such as snake venoms, two peptide toxins (waglerin-1 and -2) were first isolated from Wagler's pit viper. These peptide toxins consist of 22 amino acids each, and kill experimental animals through unknown mechanisms, but partly through nerve cell attachment via nicotinic acetylcholine receptors [18]. Synthetic peptides based on the natural toxin sequences also show similar toxicity to host cells in vivo [19]. At present, a variety of roles of peptides have been identified in many systems, including antimicrobial peptides in innate immunity and control of activation of receptors for growth factors that are involved in signaling pathways [20, 21].
Because exported substances (mainly proteins) from a cell could negatively affect other cells or the cell itself, multicellular organisms may have evolved mechanisms to avoid these phenomena. For example, apoptosis, a type of programmed cell death, produces cell fragments (apoptotic bodies) that are encapsulated and engulfed by phagocytic cells, which prevents the leakage of the intracellular contents of dead cells [22]. Autophagy is another basic catabolic mechanism ubiquitous in eukaryotic cells and serves to degrade dysfunctional cellular components within double-membrane encapsulated structures associated with lysosomes [23]. Neutrophil and eosinophil extracellular DNA traps are composed of a meshwork of DNA fibers and granule proteins that are toxic to pathogens and possibly host cells [12, 13]. One of the major functions of apoptosis, autophagy, and granulocyte (neutrophil and eosinophil) extracellular DNA traps is considered to be the prevention of leakage of toxic substances from injured cells which reduces needless inflammation [13, 22, 23].
To summarize, various sized protein derivatives, including monoamines and peptides, derived from the cells of a given host (endogenous PPs), as well as exogenous toxic proteins from pathogens, can adversely affect the cells of their origin or other cells. Because some smaller protein substances (i.e., pathogenic peptides) can be toxic to cells, there should be a control system regulating them within the host and the author proposes that T cells may control those small substances that cannot induce antibodies.
Go to :
Pathologic findings in infectious diseases
Generally, patients with a specific disease would have similar pathologic findings of injured tissues that are characteristic of the disease itself. Immune cells observed in the pathologic lesions originate from circulating immune cells from the blood (i.e., neutrophils, eosinophils, basophils, lymphocytes, monocytes, natural killer cells, etc.). The distribution of immune cells in pathologic lesions may be different according to the disease entity and the stage of the diseases. It has been thought that neutrophils and phagocytic monocytes (macrophages) are the predominant cells in early lesions of bacterial diseases, including mycoplasma pneumonia, and lymphocytes are predominant in early lesions of viral diseases, such as influenza pneumonia. However, neutrophils and macrophages are prominent in the very early stages, and lymphocyte infiltration follows rapidly in the early stages of both mycoplasma and influenza infections [24, 25]. In addition, eosinophils are the predominant cells in pathologic lesions associated with visceral larva migrans, a disease caused by a specific parasite that induces severe eosinophila and eosinophilic pulmonary lesions [26]. These findings suggest that innate immune cells may recognize the substances commonly produced in the early lesions, rather than those of a particular kind of pathogen. In addition, precipitated immunoglobulins in pathologic lesions vary according to disease entities. In renal diseases, the classes/subclasses and location of the precipitated immunoglobulins (IgG, IgG subclasses, IgA and IgM) differ according to the type of renal disease. Acute poststreptococcal glomerulonephritis (APSGN) is thought to be an immune-complex induced disease that results from Group A streptococcal infection, but many patients with the disease have no IgG deposition in renal pathology sites [27]. IgA nephropathy is a chronic renal disease characterized by IgA deposition in mesangial areas of glomeruli, but this finding is noted in other systemic diseases, including liver cirrhosis and chronic drug abuse [28]. Thus, immunoglobulins found in renal tissues may be secondary phenomena from systemic immune reactions rather than primary etiologic agents, and these phenomena could also occur in pathologic lesions in other infectious diseases.
Various pathogens, including bacteria, respiratory viruses, mycoplasmas, chlamydias, rickettsias, fungi and parasites, are known to be causative agents of pneumonia [29]. Only a subset of patients infected by the pathogens develops pneumonia and, occasionally, extrapulmonary manifestations such as skin rash, meningoencephalitis, myositis and other organ involvements can occur during these infections. However, it is unknown whether lower respiratory tract cells and extrapulmonary organ cells have all the receptors needed for entry into the cells by a wide variety of small pathogens, especially viruses. Furthermore, it is also not known whether lower respiratory tract cells are directly damaged by pathogens in vivo, because some pathogens do not exhibit cellular damage (cytopathic effects) in vitro, and only a few or no etiologic pathogens are seen in pulmonary and extrapulmonory lesions. Some rare diseases seriously damage whole organs within a relatively short period of time; these include sympathetic ophthalmia (eyes), toxic epidermiolysis (skin), fulminating hepatitis (liver), idiopathic interstitial pneumonia (lungs), acute myocarditis with dilated cardiomyopathy (heart), rapidly progressive glomerulonephritis (kidneys), acute necrotizing pancreatitis (pancreas), acute encephalopathy including Reye syndrome (brain), Waterhouse-Friderichsen syndrome (adrenal glands) and acute hemorrhagic shock syndrome (multiple organ failure) [9, 30, 31, 32, 33, 34, 35, 36, 37, 38]. Each organ-specific disease has been reported to be associated with more than one etiologic agent, including various pathogens. Although each organ-specific disease may have similar pathologic findings with massive immune cell infiltration, generally it is difficult to find whole pathogens in the extensive pathologic lesions. In contrast, there are some diseases which exhibit minimal involvement of immune cells and antibodies in pathologic lesions, including minimal change nephrotic syndrome, rotavirus gastroenteritis, rhabdomyolysis and chronic neurodegenerative disorders such as prion diseases [10, 39, 40, 41]. These findings could be explained by the actions of a defense system other than the conventional immune system that exists in vivo, and the toxic substances may be very small PPs controlled by unknown immune functions of the host that do not involve immune cells.
Although whole pathogens can be found in pathogenic lesions, the smaller substances produced in response to infectious insults may recruit immune cells and induce inflammation. Immune cells and immune proteins, including antibodies and complements, may act to control these substances according to their functional capacities in a variety of pathologic lesions.
Go to :
Etiologic agents of cell injury in infectious diseases
Clinical symptoms and signs of infectious disease, such as fever onset and various tissue injuries, are initiated by the adhesion and invasion of pathogens into a host. After invasion, pathogens replicate and induce cell destruction that provokes symptomatic illness. If microorganisms do not replicate and/or destroy tissues and cells, clinical manifestations of infectious diseases may be subclinical or very mild, even though immune responses against pathogens have been elicited. Attenuated viruses in live vaccines, such as measles-mumps-rubella (MMR) or chicken pox vaccines, may belong to this category. On the molecular biological level, the pathogens, including virions, may be too large to be directly toxic to the host cells, and thus intact pathogens may be ineffectual as large protein complexes with several surface antigens if they do not induce production of smaller substances during their replication processes.
An infectious disease usually starts as a primary infection site (the focus) where pathogens multiply and the causative substances (PPs) are produced. In extracellular bacterial infections such as scarlet fever, cellulitis and osteomyelitis, the foci of bacterial multiplication are often readily seen sites such as the tonsils, skin and bones, respectively. In contrast, with viral infections and intracellular bacterial infections such as salmonellosis (typhoid fever) and legionellosis, the initial foci for replication of viruses or intercellular bacteria are not apparent. In bacterial infections, the focus of an infection produces a lot of substances, including replicated bacteria (bacteremia), bacterial exotoxins known as superantigens, fragments of bacterial components that serve as endotoxins or present pathogen-associated molecular patterns (PAMPs), materials from activated immune cells such as proinflammatory cytokines and proteolytic enzymes, and other materials from injured cells, including damage-associated molecular patterns (DAMPs) that may be various sized newly produced proteins within the cells by the infectious insults. Therefore, systemic symptoms and signs of bacterial infection (fever, bacteremia or sepsis) may be result from the release of these substances into systemic circulation and/or nearby local regions. Smaller pathogens such as viruses, mycoplasmas, rickettsiae and chlamydias, and larger pathogens such as parasites, also invade into host tissues and establish foci, which contain replicated pathogens, exogenous smaller substances from the pathogens and endogenous toxic substances from injured host cells that may recruit corresponding immune cells. Thus, the modes of action of immune cells in these infections are believed to be similar to bacterial infections.
The use of antimicrobials is essential for elimination of susceptible pathogens and reduction of infectious insults from the infected host. However, empirical use of antimicrobials is not always advantageous for patients with infectious diseases regardless of drug resistance. For example, approximately 15% of community acquired pneumonia patients, especially older patients with underlying diseases, experience treatment failure with high mortality [42], and some patients with bacterial infection can show a transient deterioration of clinical symptoms after initial antibiotic treatment as a result of cytokine storm [43]. In mycoplasma pneumonia, antibiotic treatment induces rapid defervescence, but some patients show progressive pneumonia despite early administration of appropriate antibiotics [44]. Similar findings can be observed in influenza virus infection; early antiviral treatment induces rapid defervescence, but some patients can progress to acute respiratory distress syndrome (ARDS) with early antiviral treatment [45, 46]. For these patients, the author's group and other study groups have observed that early systemic immune modulators (corticosteroids and/or high-dose intravenous immunoglobulin, IVIG), together with antibiotics or antivirals, can prevent rapid progression of pneumonia and induce rapid recovery of pulmonary lesions in M. pneumoniae pneumonia or 2009 H1N1 pandemic influenza pneumonia [46, 47, 48, 49, 50]. In addition, corticosteroids have been used as an adjuvant treatment for many other infectious diseases, including severe bacterial infections such as typhoid fever and tuberculous meningitis [51]. Corticosteroids may act on hyperactive immune cells that are needed for disease control but they may overproduce the immune substances such as proinflammatory cytokines. The immune cells affected by corticosteroids, especially nonspecific immature T cells, B cells and eosinophils, may be rapidly eliminated by apoptosis while in circulation [52]. Corticosteroids (hydrocortisone) and IVIG can be regarded as host-origin controllers but are not produced in adequate doses within the short duration of exposure to acute massive infectious insults. Thus, the beneficial effects of immune modulators could be explained in this way, since the host immune/repair system should still be able to control all insults from an infection even if the immune modulators do not kill pathogens. The limited effect of antimicrobials and the effectiveness of immune modulators on many infectious diseases also suggest that etiologic agents inducing inflammation in pathologic lesions are not whole pathogens, but are other substances inducing immune cell activation. Kikuchi-Fujimoto disease of unknown etiology is characterized by lymphadenitis, prolonged fever and other systemic symptoms such as fatigue and weight loss [53]. The author has previously proposed that substances inducing clinical symptoms of this disease may be derived from involved lymph nodes, because removal of the affected lymph nodes induces prompt improvement of clinical symptoms (defervescence) [54].
Specific antibodies (IgM and IgG) against pathogens, including viruses, mycoplasmas and legionellas, are not detected for at least 3-4 days after the onset of clinical symptoms. Since each pathogen, especially viruses, is known to attach only to specific receptors on host cells, viruses that spread by systemic circulation (viremia) may not bind other organ-specific host cells that lack these receptors. If the virus-receptor binding and subsequent tissue cell destruction by intracellular replication needed to happen, then a period of incubation following virus infection would be required. Furthermore, in experimental animal studies of mycoplasma or influenza virus infections, animals with depressed immune functions, including those with T cell deprivation or IL-17 deficiency, show milder or fewer pneumonia lesions, compared with immune-competent animals, although duration of survival of the pathogens is longer [25, 55, 56, 57]. Therefore, the immunological reaction of host immune cells against the substances produced from the foci of pathogen replication may be responsible for lung injury, and the substances may have a similar mode of binding to lung tissue cells and recruiting the corresponding immune cells [1, 2, 3, 4] (Fig. 2).
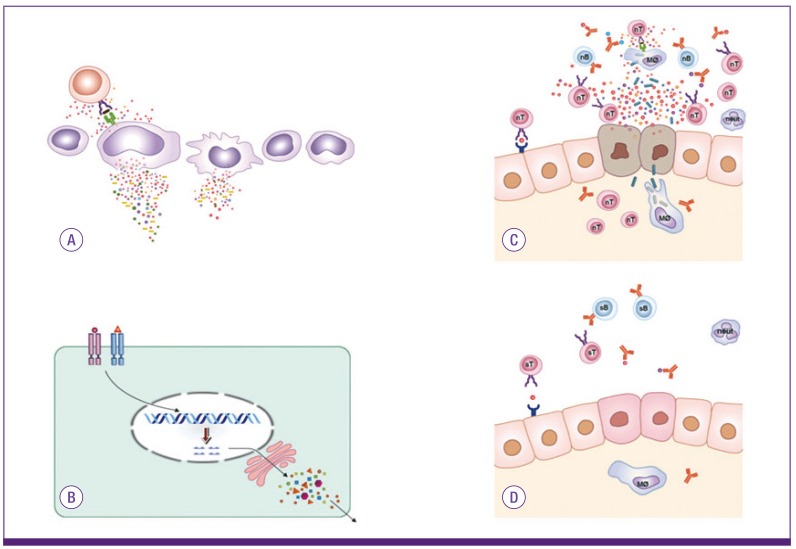 | Figure 2Immunopathogenesis of infectious diseases and infection-related immune diseases.The hosts have an initial focus in which pathogens encounter and replicate in infectious diseases or infection-related immune diseases. Various substances, including pathogens and fragments of pathogen-origin, cytokines from immune cells and materials from destructed host cells are preformed in the initial focus and/or secondary immune organs around the focus (A). These substances spread and reach various tissues via systemic circulation, and some of these bind to receptors on specific organ cells. Immune cells start to control these substances, and clinical symptoms and signs begin to appear. The pathogenic proteins (PPs) bind to receptors of target cells of the host, and this process signals cell injury and/or other protein production from target cells (B). Immune cells are recruited to the lesions for control of substances, including PPs through MHCs and cytokine networks. Initially, immune cells, including non-specific T cells and non-specific antibodies, are involved in this reaction. During this process, hyperactivated immune cells produce various inflammatory cytokines and counter-inflammatory cytokines, and the cytokine imbalances may be associated with further target cell injury (C). After appearance of specific T cell clones and B cell clones (specific antibodies) for PPs, tissue injury ceases and repair begins with immune cells (D). While the hosts that have a defect on the appearance of specific immune cells (lack of the repertoire of specific immune cells) against PPs may ensue autoimmune diseases.
|
There are many acute and chronic (autoimmune) infection-related immune-mediated diseases, including APSGN, acute rheumatic fever, KD and possibly rheumatoid arthritis. The mechanisms of immunopathogenesis of these immune-mediated diseases are also not completely known, although it has been proposed that substances such as exotoxins derived from pathogens elicit direct destruction of organ specific tissue cells, or that adaptive immune cells (specific T cells and/or tissue specific autoantibodies) attack those host tissue cells that share epitopes with infectious agents (molecular mimicry) [58, 59, 60]. In the PHS hypothesis, the initial and hyperimmune response of non-specific immune cells (T cells and non-specific antibodies) against PPs may be responsible for cellular damage, and specific T cells and specific antibodies for PPs may eliminate the causes of the diseases. APSGN and acute rheumatic fever are representative infection-related immune-mediated diseases that occur after an infection with known pathogens, mainly group A β-hemolytic streptococci (GAS). However, GAS or structural components of GAS are not observed in renal lesions of APSGN or in heart lesions of acute rheumatic fever [58, 59], and the same absence of pathogen-derived substances in lesions of host tissue may be true of most of infection-related immune-mediated diseases. For recovery from an initial GAS infection, host immune cells should control all kinds of substances of the initial inflammation, including those from the pathogens and substances from immune cells and injured host cells if they are toxic to the host cells. During this process, some PPs are produced and attach primarily to kidney cells or to heart cells during the incubation period between the initial GAS infection and the onset of APSGN or rheumatic fever. Activation of non-specific immune cells against PPs, including activation of the alternative complement system, may be responsible for the inflammation and clinical symptoms. After formation of the specific T cells and specific antibodies that effectively control the PPs, the disease process halts, and the regeneration response of immune cells begin (Fig. 2).
As for each immune-mediated disease, only hosts with genetic defects in controlling the PPs produced from initial infection may be affected by the disease, and the respective affinity of PPs to host target cells, with subsequent corresponding immune cell responses, may define autoimmune disease phenotypes. For example, the main target cells in KD, rheumatoid arthritis and type 1 diabetes mellitus may be the coronary artery endothelial cells, the chondrocytes of joints, and the pancreatic β-cells, respectively [5]. The severity or chronicity of infectious diseases and immune-mediated diseases depends on the amount of PPs and the duration of appearance of specific immune cells or the repertoire of specific immune cells that can control PPs in the host. As well as in sympathetic ophthalmia, it is possible that some PPs in acute or chronic organ-specific diseases, including idiopathic interstitial pneumonia, fulminating hepatitis, myocarditis and other diseases as previously described, originate from the organ specific target cells which are injured by initial PPs from the primary focus of infection.
Go to :
Immune responses against infectious insults from various pathogens
The author previously proposed that the basic function of immune cells may be to recognize and control toxic substances of specific sizes and characteristics [2, 3, 4, 5]. Briefly, larger protein complexes, such as whole pathogens (bacteria and viruses) and large pieces of destroyed cells (apoptotic or necrotic debris), are eliminated by natural immune system cells (e.g., neutrophils and phagocytic monocytes). B cells control medium-sized proteins via the production of antibodies, while T cells control small proteins (peptides) via secretion of cytokines, possibly T cell receptor (TCR) gene-related products against these peptides or by the effects on the peptides when they bind to cells (Fig. 3). Since smaller peptides, such as monoamines and neurotransmitters, cannot be controlled by T cells with TCRs, other control systems may exist in vivo, as previously suggested.
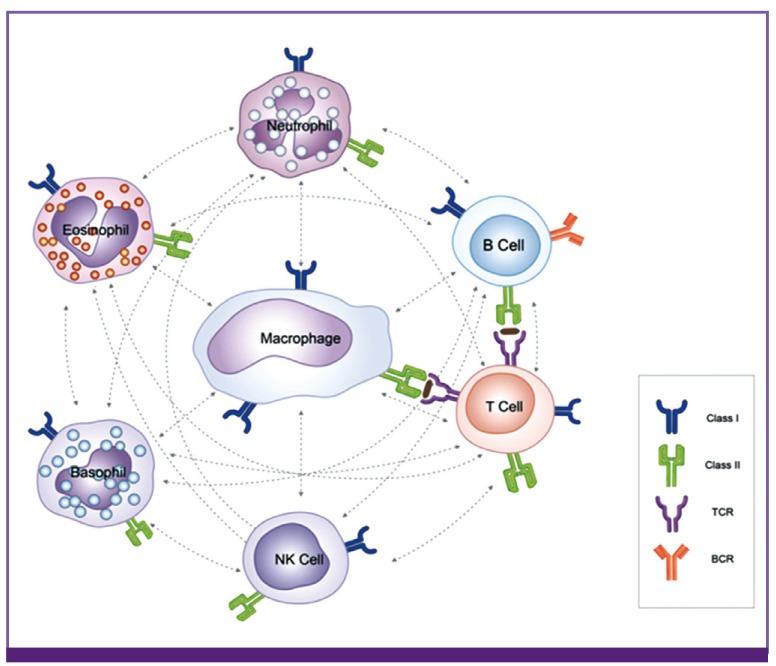 | Figure 3Immune cells as one part of the PHS.Immune system in mammals consists of various immune cells. Since circulating immune cells may be only effectors for tissue cell protection and tissue cell repair in most pathologic lesions, each immune cell may have its characteristic roles for control of substances that are harmful against host cells. Although each immune cell has a variety of biological functions, its basic function is thought to recognize the sizes and characteristics of toxic substances, including proteins, and control them. For simple examples, neutrophiles and phagocytic monocytes control larger protein complexes, such as whole pathogens and large pieces of destructed cells (apoptotic or necrotic debris). Eosinophils may control substances from parasites by secretion of proteins in granules of the cells, and natural killer cells control damaged cells or tumor cells via proteins within the cells. Adaptive immune cells control small protein substances; B cells control medium-sized proteins via production of antibodies, while T cells control small proteins (peptides) that cannot induce antibodies. Macrophage-linage cells play the most important roles in immune/repair mechanism of the host, and possibly control the small non-protein substances such as lipopolysaccharides, viral RNAs and DNAs (PAMPs) through cell receptors including Toll-like receptors, as well as natural autoantibodies do against non-protein substances. In any systemic and micro-environmental insults from a given infection, immune cells may communicate each other via MHCs, costimulatory ligands and cytokine networks to recover the adverse circumstances under the control of PHS. Through these functions, immune cells participate in control of toxic substances, including exogenous PPs from various pathogens and endogenous PPs from destructed tissue cells, and they also participate in the reconstruction of damaged tissue.
PHS, protein-homeostasis-system; TCR, T cell receptor; BCR, B cell receptor; NK cell, natural killer cell.
|
In this section, the author discusses the unsolved immunological issue in which T cells control the levels and activities of peptides, including pathogenic peptides. Although the mammalian immune system has been categorized into innate (or natural) and adaptive (or specific) immune systems, only the innate immune system exists for insects and plants, which have existed for a much longer geologic period than mammals have. In the innate immune system, the toll receptors of fruit flies and the toll-like receptors (TLRs) of mammals have similar structures and mechanisms of action against invading pathogens [61]. Thus, the innate immune system still plays a significant role in vertebrates, and the adaptive immune system has appeared much later than the innate system, with some exceptions [62]. However, the cells of these two immune systems react mutually against any external or internal insults because both types of immune cells are found in a variety of pathological lesions. Among immune cells, the macrophage-lineage cells are believed to be the most crucial component of the overall the immune system, because these cells appear critical in embryonic development and have omnipotent roles including immune reaction, tissue regeneration and hormone production (Fig. 3) [63, 64].
In the adaptive immune system, the mechanism of recombination of various immunoglobulin genes in B cells to cover the extensive antigenic diversity of proteins was first described [65], followed by the discovery that T cells also use a TCR gene recombination mechanism to accommodate the diversity of peptide antigens [66]. The roles of B cells (antibodies) are diverse and have been studied in various fields, including the prevention of infectious disease (vaccine study), autoimmune diseases (immune-complex diseases), monoclonal antibodies for diagnostics and therapeutics and IVIG treatment. In contrast, T cells have been regarded as a mainstay of adaptive immunity of almost all infectious diseases and immune-mediated diseases, but known functions are extremely limited, but do include the involvement in antigen presentation with MHCs on antigen presenting cells (APCs), a helper role for antibody production and the cytotoxic effect toward virus-infected cells. Therefore, it is possible that unknown functions of T cells could be inferred by analyzing the roles of B cells.
B cells can produce antibodies that bind to any proteins recognized by B cell receptors (BCRs), but they cannot induce antibodies against small foreign proteins (peptides) and the majority of serum proteins, except intracellular proteins. Although it is believed that B cells cannot produce antibodies of their own, some healthy persons without any symptoms have various autoantibodies against proteins from various host tissue cells, as do patients with autoimmune diseases such as systemic lupus erythematosus [67]. After infection with a pathogen, a variety of pathogen-specific antibodies against the pathogen are thought to be produced by B cells (plasma cells). A given plasma cell clone has a kind of BCR for only a certain-sized protein as an antigen and produces a specific antibody against the protein. Thus, the antigens that induce pathogen-specific IgG antibodies are not the intact pathogens (virions, bacteria, and large complex proteins), but the fragments of structural proteins and/or other proteins from gene products of pathogens. Among numerous pathogen-specific antibodies, only some specific IgM and IgG antibodies may bind to the pathogens in vitro and in vivo, and have been used as diagnostic tools. In a virus infection such as mumps, several serologic methods for diagnosis have been developed, including serum neutralization, complement fixation, hemagglutination-inhibition, immunofluorescence and enzyme immunoassays [68]. Since the antigens used in each diagnostic tool differ slightly according to the production methods and specific laboratory protocols, the positive rates of pathogen-specific antibodies in each method may be different [69]. Recently, a large number of mumps outbreaks in two-dose MMR vaccinees have occurred globally, including in Korea [4, 70]. Almost all of the two-dose vaccinated patients are anti-mumps IgG positive at the beginning of the illness [4, 71]. Therefore, in mumps virus infection, waning immunity over time is not reflected by the vaccine-induced IgG antibodies [4]. Similar findings are observed in other viral infections such measles and chicken pox, and vaccinees can be infected over time after vaccination [72, 73]. In the 2009 H1N1 virus pandemic, the pre-pandemic seroprevalence of neutralizing IgG antibodies that were cross-reactive to the H1N1 influenza viruses did not match those of susceptible age groups in the general population, including in Korea, suggesting that older people could have pre-existing immunity not detectable by cross-reactive IgG antibodies [74, 75]. These findings suggest that not only pathogen specific IgG but also other immune substances (possibly specific immune peptides?) are necessary for prevention of virus attachment and replication in the host.
The level of immunoglobulins (IgG, IgA and IgM) is different between individuals, but the level of IgG may remain relatively constant in the healthy state of each adult individual. It has been noted that serum immunoglobulins consist not only of antibodies for pathogens (specific antibodies) but also of antibodies, which control the immune reaction in vivo (natural antibodies). Natural antibodies are produced by B1 type B cells in the absence of external antigenic stimuli and are already present in the fetus and the newborn at birth [76, 77]. Thus, it has been suggested that they are involved in innate immune reactions against pathogens before the establishment of adaptive immunity. Additionally, these antibodies, especially IgM antibodies, have the ability to bind to various self and foreign substances, including non-protein substances such as oligosaccharides, polysaccharides, nucleotides and phospholipids. These findings suggest that the main function of natural antibodies may be to control various substances originating from self-cells, including non-protein materials, although they may have other immune regulatory functions in vivo [76, 77]. The author and colleagues observed that high-dose IVIG decreased all serum protein levels temporarily except for immunoglobulins (IgA and IgM) in KD patients [78, 79], and that there was a positive relationship between low albumin and low IgG values in children with idiopathic steroid-sensitive (minimal change) nephrotic syndrome in which urinary loss of IgG is nearly absent [80]. These findings support the hypothesis that IgG recognizes proteins in the serum and controls the proteins systematically, and it provides an important component of the foundation for the PHS hypothesis [81]. Therefore, it could be inferred that the common action of B cells is to control the activity of non-self exogenous substances, mainly proteins and some endogenous substances, including PPs, which find their way into systemic circulation.
It is believed that the TCRs of T cells, together with MHC molecules, can control all antigenic peptides as a result of recombination of TCR genes. The length of peptides recognized by T cells is not consistent, but there is only a limited range of peptides consisting of ~12-25 amino acids that cannot induce antibodies. In the PHS hypothesis, it is possible that T cells may produce specific peptide-control substances associated with TCR genes or other products such as specific cytokines which can control specific peptides, not unlike how B cells produce specific antibodies recognized by BCR and maintain a serum level of specific IgGs.
MHC class I molecules are expressed by nearly all mammal cells, while MHC class II molecules are expressed principally in immune cells and show individual polymorphisms [82]. MHC molecules may be one of the markers of self-identification and may be used for communication between circulating immune cells. Thus, immune cells with different MHCs do not respond to (or communicate with) each other in certain immune reactions, the so-called MHC restriction phenomenon [83]. In the PHS hypothesis, T cells regulate peptide binding to various tissue cells, including those peptides that bind within class I MHC molecules, and T cells can be activated by these peptides. It is possible that MHC class I molecules distributed on all tissue cells act as receptors for circulating exogenous and endogenous peptides. Heat shock proteins (HSPs) are unique proteins that function as molecular chaperones to prevent misfolding and aggregation of nascent polypeptides and facilitate normal protein folding [84]. Some HSPs in the cytosol and endoplasmic reticulum can bind antigenic peptides generated within the cells. The antigenic peptides that are chaperoned by HSPs are transported to the MHC class I molecules on the cell surface for presentation to T cells. In addition, extracellularly released chaperoned peptides can be taken up by APCs via receptor-mediated endocytosis, whereupon they are presented to T cells via MHC class I molecules on the surface of APCs, a process called cross-presentation. The HSP-peptide complexes acting through cross-presentation induce stronger T cell activation, compared with antigenic peptides alone [85]. Therefore, it is possible that circulating pathogenic peptides, including HSP-peptide complexes from pathogens and injured cells, could be directly captured in MHC molecules or other receptors of the host cells. These peptides could activate T cells as well as the peptides presented by APCs via the classical antigen presentation pathway. As previously mentioned, some peptide proteins such as snake venoms (waglerin-1 and -2) can directly affect host cells [18, 19]. Various MHC polymorphisms in individuals may be considered to reflect the repertoire of peptides that can be controlled together with TCRs in each individual. Therefore, the association between certain disease frequency and MHC polymorphism could be explained by this assumption. Furthermore, antibodies for different MHCs exist in multipara and multi-transfused patients, and MHC polymorphism in individuals can be classified by serologic methods [86]. Thus in organ transplantation, self-tissue MHC proteins are recognized by the PHS as self-markers so that there are no immune responses. Foreign-tissue MHC proteins and other numerous structural antigenic proteins from other individuals or animals (external PPs) would be attacked by immune cells together with nonspecific antibodies, and foreign peptides within MHCs or possibly other receptors on foreign cells would also be attacked by non-specific T cells. The concepts of self and non-self discrimination of the immune system has changed, leading one immunologist to propose that initiation of the immune response may be associated with substances from cell necrosis that induce APC activation regardless of self and non-self discrimination (the Danger model) [87].
It has been proposed that proteins produced by the innate immune system affect adaptive immune cells. In the initiation of an infection, the fragments of pathogens (PAMPs), including bacterial lipopolysaccharides (LPS), viral DNAs and viral RNAs, bind the respective ligands (pattern recognition receptors, PRRs) such as TLRs and intracellular sensors (i.e., nuclear oligomerazation domains, etc.) in macrophages or in the infected cells [88, 89]. The PAMP-PRR binding produces anti-pathogenic proteins (in the case of viruses, interferons and related proteins) and proinflammatory cytokines via the receptor-signal transduction system. It was postulated that these proteins may be associated with the functions of adaptive immune cells, such as their expression of costimulatory receptors or the differentiation of T cell subtypes [88, 89]. However, it could also be postulated that these receptors may be partly responsible for independent control of small non-protein toxic substances such as PAMS, by innate immune system cells, whereas BCRs or TCRs work against protein toxic substances (i.e., PPs) by adaptive immune system cells, since these receptor-antigen bindings do cause receptor signal transduction that results in production of new proteins for controlling pathogens, such as anti-pathogen proteins or pathogen-specific antibodies. Additionally, natural antibodies that can bind non-protein substances from pathogen or self-cell origin could be regarded as part of the innate immune system [76, 77]. The complement system, one of innate immune system produces immune peptides such as C3a and C5a (anaphylatoxins) during immune reaction, although their functions are not fully understood [90]. Again, the immune system of a host could control intact pathogens macroscopically (via phagocytosis), and fragments of the pathogens, including proteins and non-protein materials, microscopically (Fig. 3).
T cells, together with other immune cells, appear early in pathologic lesions of nearly all human diseases, including infectious diseases, rheumatic diseases, cancers, transplantation rejection and regeneration of tissues (keloids). This finding suggests that T cells are involved in pathophysiologic phenomena in various disorders, and the modes of their actions may not be different across various disorders. Those T cells that appear in early pathologic lesions may be non-specific naive T cells, which do not recognize the presented peptides from APC cells. When exogenous or endogenous PPs bind to the receptors of target cells, signals from the affected cells recruit immune cells, and proteins from injured cells also recruit corresponding immune cells for clearance of exposed substances including possible PPs. In early inflammatory sites, innate immune cells, non-specific T cells and B cells and non-specific antibodies (and possibly non-specific immune peptides) and other immune proteins (complements) constitute the first-line effectors for control of inflammatory mediators, although they are not more effective than specific effectors. Following communication between these immune cells, the hyperactive immune phenomena, including overproduction of cytokines, may be responsible for injury of certain tissue cells in severely affected patients. Eventually, inflammation ceases after the appearance of specific immune cells (T cells) with specific antibodies (B cells) that effectively control protein substances, including PPs. Therefore, if a host's PHS could not control the PPs and proteins from pathogens and injured cells, a disease would progress to the death of the host in the case of acute infections, or cause chronic autoimmune disease due to the continuous activation of non-specific immune cells (Fig. 2).
Among primary immunodeficiencies, patients with B cell deficiency have minimal amounts of IgG and other immunogobulins, and most cases experience mainly bacterial infections with relatively good prognosis if given timely antibiotic treatment and periodic IVIG infusion. In contrast, patients with severe T-cell deficiency have low levels of IgG, owing to the lack of communication between the immune cells (T-B cell interaction), and these patients suffer from serious infections caused by various pathogens, including most viruses, bacteria, fungi and protozoa, with eventual fatal outcomes [91, 92]. In the in vitro setting, the T cells may not directly suppress or kill such pathogens, unlike phagocytes and pathogen-specific antibodies. At least a partial explanation could be that immune substances that are produced from T cells, and are needed to suppress pathogen replication, are lacking, and/or the pathogenic peptides derived from infectious insults cannot be controlled by T cells, and thus the damage to tissue cells is ongoing. This reasoning would also explain why partial T-cell deficiencies are associated with higher risks for autoimmune diseases [92].
Since T cells can be activated using various mitogens and monoclonal antibodies against the cell membrane receptors in vitro, various activation mechanisms of T cells are expected in different pathologic lesions in vivo. For example, superantigens bind to the sides of TCRs and MHC class II molecules, and induce cytokine production from numerous activated T cells. Although hypercytokinemia from activated T cell clones is believed to be responsible for diseases such as toxic shock syndrome, it is unknown whether superantigens simultaneously bind MHCs and TCRs in vivo, and there are many other receptors for superantigens on the host cells [93, 94].
T cells have been classified by the presence of cell surface antigens, such as CD4+ helper T cells and CD8+ cytotoxic T cells. Helper T cells are classified into Th1, Th2, Th17, and regulatory T cells (Treg cells) depending upon the types of cytokines expressed. It is proposed that the balance of these T cell subsets may be important for maintenance of the immunologic homeostasis of a host. However, this pattern is not predictable across various diseases and even across instances of the same disease [95, 96]. Thus, various T cell subsets are expected to change phenotypes and secrete different cytokines that correspond to pathogenic peptides in pathologic lesions [97]. Additionally, specific T cell subgroups, such as γδ T cells or innate helper cells, exist in certain organs in mammals. These T cells are activated and secrete cytokines when the respective organ cells are injured, suggesting an involvement in tissue repair [98]. It was also reported that T cells may be involved in lymphangiogenesis in lymph nodes through cytokines such as INF-γ as well as B cells and macrophages [99, 100].
To summarize, T cells have various roles in the homeostasis of a healthy state in mammals. The concept that T cells control small proteins (pathogenic peptides) may be essential to explain the integration of the T cell's roles in infectious diseases, although this remains to be proven in the future.
Go to :
Conclusion
All biological activities of an organism, including immunological responses to infectious insults from pathogens, are carried out by proteins encoded by the toolbox of genomic DNA. There are numerous small protein derivatives in organisms, including monoamines and peptides, and the activities of these substances are strictly controlled by the in vivo system. Therefore, organisms may recognize the self-structural and functional proteins, eliminate those proteins harmful against the host cells, supply alternative proteins for deficiency of a protein and maintain the protein homeostasis for a healthy state of the organism. Given that circulating immune cells and immune proteins (antibodies, complements, cytokines and possibly immune peptides) are the only effectors for prevention of tissue cell injury of the host, it is reasonable that immune cells recognize and act against the wide range of sizes and properties of the substances, including PPs and pathogenic peptides.
Go to :
 XML Download
XML Download