

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abstract
Primary sclerosing cholangitis (PSC) is a slowly progressive cholestatic liver disease. In cases of PSC, liver transplantation is the only effective treatment that can delay the disease’ s natural course. We report a case of rapidly progressive PSC requiring liver transplantation. A 52-year-old woman visited our hospital with abdominal pain. There was no evidence of PSC, as there was no elevation in cholestatic liver enzymes at her first visit. Although her total bilirubin was in a normal range at the initial visit, liver dysfunction progressed rapidly. Despite endoscopic procedures and ursodeoxycholic acid intake, total bilirubin levels rose to 18.9 mg/dL, and liver transplantation was performed 17 months after her first visit. PSC was pathologically confirmed after liver transplantation.
Go to : 
REFERENCES
1). Molodecky NA, Kareemi H, Parab R, Barkema HW, Quan H, Myers RP, et al. Incidence of primary sclerosing cholangitis: a systematic review and meta-analysis. Hepatology. 2011; 53:1590–9.

2). Milkiewicz P, Wunsch E. Primary sclerosing cholangitis. Recent Results Cancer Res. 2011; 185:117–33.
3). Rosen CB, Nagorney DM, Wiesner RH, Coffey RJ Jr, LaRusso NF. Cholangiocarcinoma complicating primary sclerosing cholangitis. Ann Surg. 1991; 213:21–5.
4). Broomé U, Olsson R, Lööf L, Bodemar G, Hultcrantz R, Danielsson A, et al. Natural history and prognostic factors in 305 Swedish patients with primary sclerosing cholangitis. Gut. 1996; 38:610–5.
5). Tischendorf JJ, Hecker H, Krüger M, Manns MP, Meier PN. Characterization, outcome, and prognosis in 273 patients with primary sclerosing cholangitis: a single center study. Am J Gastroenterol. 2007; 102:107–14.
6). Mehal WZ, Lo YM, Wordsworth BP, Neuberger JM, Hubscher SC, Fleming KA, et al. HLA DR4 is a marker for rapid disease progression in primary sclerosing cholangitis. Gastroenterology. 1994; 106:160–7.
7). Farrant JM, Doherty DG, Donaldson PT, Vaughan RW, Hayllar KM, Welsh KI, et al. Amino acid substitutions at position 38 of the DR beta polypeptide confer suscept-ibility to and protection from primary sclerosing cholangitis. Hepatology. 1992; 16:390–5.
8). Olerup O, Olsson R, Hultcrantz R, Broome U. HLA- DR and HLA-DQ are not markers for rapid disease progression in primary sclerosing cholangitis. Gastroenterology. 1995; 108:870–8.
9). Chapman R, Fevery J, Kalloo A, Nagorney DM, Boberg KM, Shneider B, et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology. 2010; 51:660–78.
10). Burak KW, Angulo P, Lindor KD. Is there a role for liver biopsy in primary sclerosing cholangitis? Am J Gastroenterol. 2003; 98:1155–8.
11). Baluyut AR, Sherman S, Lehman GA, Hoen H, Chalasani N. Impact of endoscopic therapy on the survival of patients with primary sclerosing cholangitis. Gastrointest Endosc. 2001; 53:308–12.
12). Gluck M, Cantone NR, Brandabur JJ, Patterson DJ, Bredfeldt JE, Kozarek RA. A twenty-year experience with endoscopic therapy for symptomatic primary sclerosing cholangitis. J Clin Gastroenterol. 2008; 42:1032–9.S.
13). Stiehl A, Rudolph G, Klöters-Plachky P, Sauer P, Walker S. Development of dominant bile duct stenoses in patients with primary sclerosing cholangitis treated with ursodeoxycholic acid: outcome after endoscopic treatment. J Hepatol. 2002; 36:151–6.
14). Shi J, Li Z, Zeng X, Lin Y, Xie WF. Ursodeoxycholic acid in primary sclerosing cholangitis: meta-analysis of randomized controlled trials. Hepatol Res. 2009; 39:865–73.
15). Poropat G, Giljaca V, Stimac D, Gluud C. Bile acids for primary sclerosing cholangitis. Cochrane Database Syst Rev. 2011; (1):CD003626.
16). Triantos CK, Koukias NM, Nikolopoulou VN, Burroughs AK. Meta-analysis: ursodeoxycholic acid for primary sclerosing cholangitis. Aliment Pharmacol Ther. 2011; 34:901–10.
17). Lindor KD, Kowdley KV, Luketic VA, Harrison ME, McCashland T, Befeler AS, et al. High-dose ursodeox-ycholic acid for the treatment of primary sclerosing cholangitis. Hepatology. 2009; 50:808–14.
18). Imam MH, Sinakos E, Gossard AA, Kowdley KV, Luketic VA, Edwyn Harrison M, et al. High-dose urso-deoxycholic acid increases risk of adverse outcomes in patients with early stage primary sclerosing cholangitis. Aliment Pharmacol Ther. 2011; 34:1185–92.
Go to :
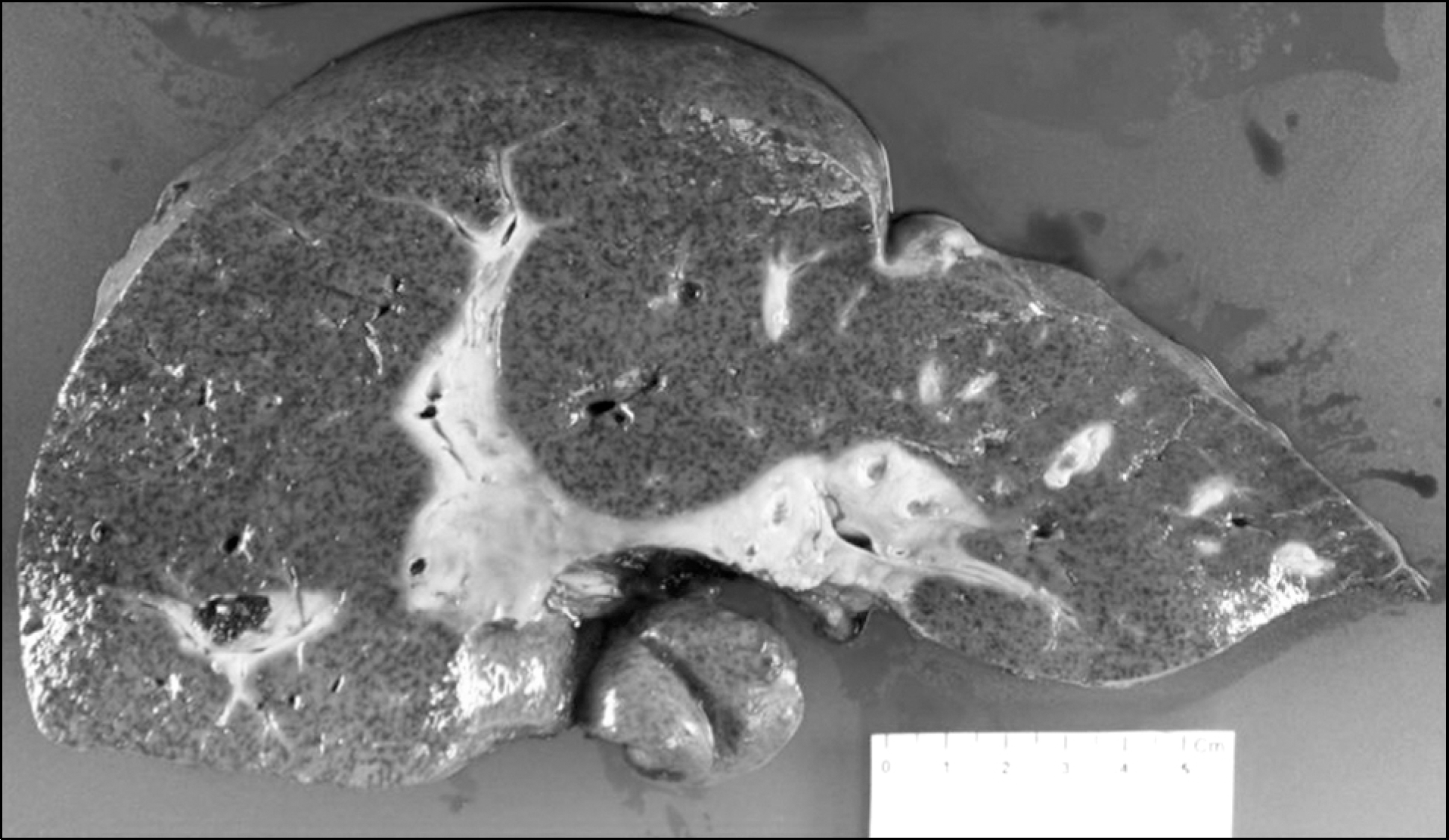
 | Fig. 1.(A) Eight months after the initial diagnosis of primary sclerosing cholangitis (PSC), the patient’ s liver biopsy showed a mild degree of lobular and portal activity, and portal fibrosis suggestive of chronic hepatitis (HE stain, ×400). (B) After liver transplantation, which was done 17 months after the initial diagnosis, surgical pathology showed periductal lamellar fibrosis, called “ onion-skin,” a typical finding of PSC (HE stain, ×400). |
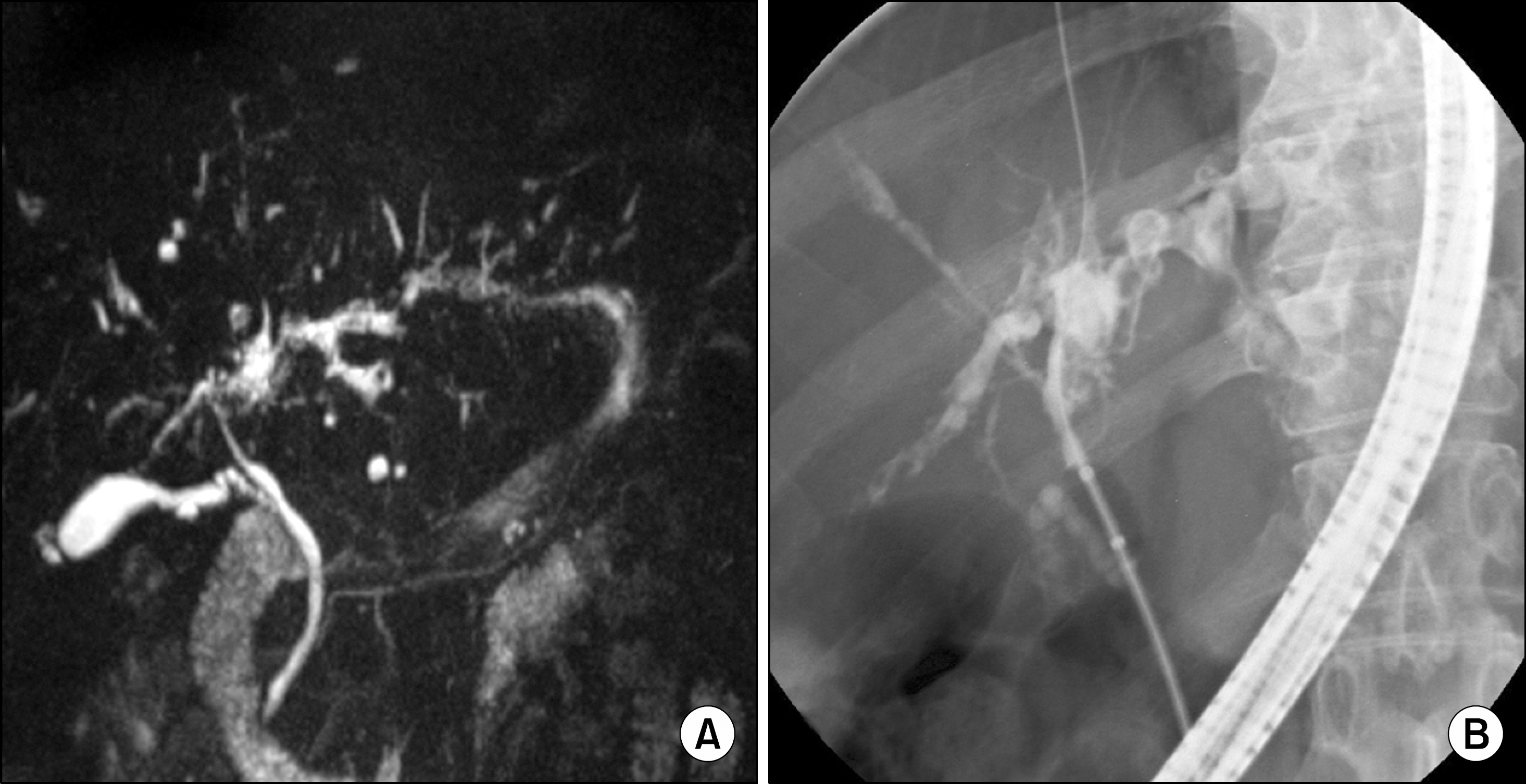 | Fig. 2.Twelve months after the initial diagnosis of primary sclerosing cholangitis (PSC), (A) magnetic resonance cholangiopancreatography and (B) endoscopic retrograde cholangiopancreatography findings showed multiple dilatations and strictures of intrahepatic ducts, suggestive of PSC. |
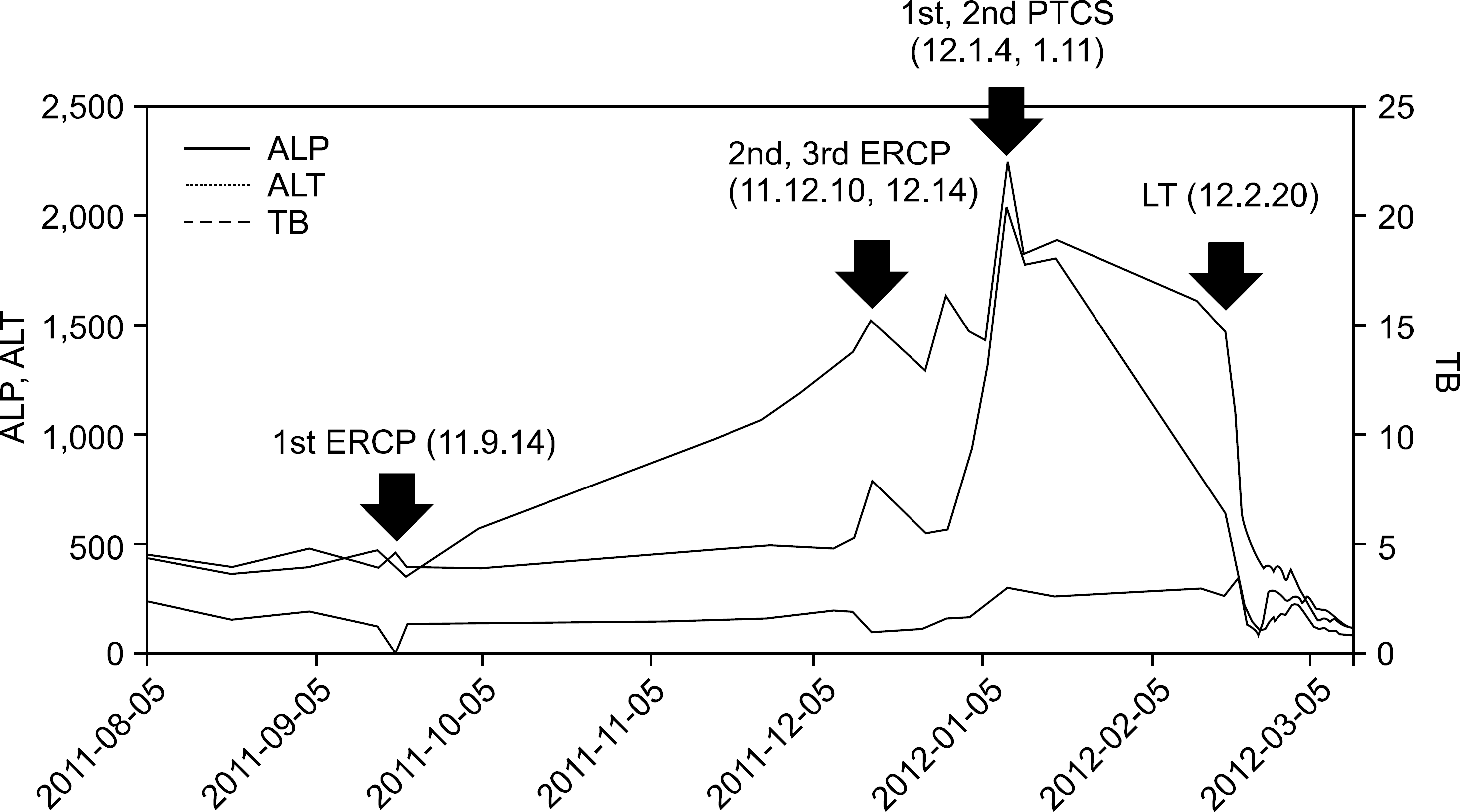 | Fig. 3.The trend of total bilirubin, alkalaine phosphatase, and alanine aminotransferase (ALT) was descri-bed. Abbreviations: PTCS, percuta-neous transhepatic choledochoscopy; ERCP, endoscopic retrograde chol-angiopancreatography; LT, liver tra-nsplantation; ALP, alkaline phosphatase; TB, total bilirubin. |
 XML Download
XML Download