

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Viruses are small infectious agents that replicate only within the living cells of other organisms and can infect all types of organisms, from plants and animals to microorganisms, including bacteria and archaea (1234). Because of the rapid increase in viral taxonomy studies, a vast number of viruses are classified among 4404 species, 735 genera, 122 families (including 35 subfamilies, and eight orders (https://talk.ictvonline.org/taxonomy/). In fact, viruses are classified by the International Committee on Taxonomy of Viruses system and the Baltimore classification system mainly according to several phenotypic characteristics. The Baltimore classification depends on the genome type (DNA, RNA, single-stranded (ss), double-stranded (ds), etc.) and replication method. According to the Baltimore classification, viruses are divided into seven classes, as shown in Table 1 (5).
As mentioned above, viral growth and multiplicity depend on a living host. Thus, virus-induced diseases occur only when pathogenic viruses enter the host's body after entering the infectious virus particle to the host's cells (6). Most but not all pathogenic viruses can cause cytopathic effects in the host cell and sometimes cause death of the host cell (7). Some viruses do not cause changes in host cells and are latent and inactive. However, viruses such as the Epstein-Barr virus (EBV) and papillomaviruses often cause cell proliferation or cancer without malignancy (89).
Viruses often cause “minor” illnesses compared with bacterial or fungal ones, e.g., colds, coughs, flu, sore throat, chickenpox, and other rashes (10). The disease commonly known as “the flu” is an infectious disease caused by an influenza virus. However, influenza viruses often lead to the development of pandemics or outbreaks. The varicella zoster virus (VZV), called the chickenpox virus, is associated with various symptoms. The VZV mainly multiplies in the lungs and, when reactivated, causes neurological conditions such as encephalitis. There are also latent viruses that are in a latent state in cells but are pathogenic, such as the cytomegalovirus (CMV) and the EBV (1112). Some viruses that are not latent cause chronic or lifelong infection (1314). Despite the host defense mechanisms, the virus continues to replicate in the host's body, and finally causes chronic disease. In particular, the hepatitis B virus and hepatitis C virus cause chronic hepatitis by inducing liver inflammation in humans (15). Other viruses cause cancer or serious illnesses with a high mortality rate. The human papillomavirus (HPV) is the most common of these viruses, causing cervical cancer. Although most people with HPV infection do not develop cervical cancer, some types of HPV, such as types 16 and 18, represent a major risk factor for cervical cancer (16). The Ebola virus has a high mortality rate (50~89% depending on the virus subtype) and causes viral hemorrhagic fever in humans (17).
The diagnosis of viruses that cause disease in humans can be generally divided into serological methods, virus antigen detection, virus culture, and viral nucleic acid detection (18192021). In recent years, the real-time polymerase chain reaction (PCR) technology method for viral genome detection has been used widely in the diagnosis of viruses because of its relatively high accuracy for specific target DNA/RNA and because it allows high-throughput quantification (222324). Real-time PCR is used widely, not only for viral detection but also for the identification of genetic mutations related to viral resistance and for virus-type classification (252627).
In this manuscript, we review virus diagnostics, especially the types of real-time PCR in molecular biology diagnostics, the issues that should be taken into consideration during such analyses, and methods that can be used to evaluate performance.
1. Diagnostic tests for viral infection
The diagnosis of viral infections can be generally performed by viral culture, serological tests, virus antigen detection, and viral nucleic acid detection.
1.1 Viral culture
The culture method is the “gold-standard” approach to the diagnosis of viruses that can be cultured. When growing a virus using cell culture, virus-infected cells exhibit morphological changes that often depend on the characteristics of the specific virus type. The virus culture can identify infection with the following live viruses: adenoviruses, cytomegaloviruses, enteroviruses, herpes simplex viruses, influenza viruses, parainfluenza viruses, rhinoviruses, respiratory syncytial viruses, varicella herpesviruses, measles, and mumps (18). Although it uses the live virus from clinical samples, the confirmation of the results of the culture method takes more than 5~9 days; moreover, it is difficult to diagnose the viral infection early and to identify virus type when using this method (282930). Therefore, this method is not usually used in clinical settings but only in the laboratory.
1.2 Serological tests
Serological tests are used to identify antibodies and antigens by diagnosis using antibodies that react specifically with the virus in question; i.e., the serological diagnosis is established by demonstrating the presence of viral-specific IgM in the sera collected at the acute phase of the disease or by proving that the viral-specific IgG titers in the recovery serum are four fold or greater rise than those observed in the acute phase (31). The serological diagnosis of viral infections has traditionally been performed via neutralization, complement fixation, hemagglutination inhibition, immunofluorescence, and enzyme-linked immunosorbent assay (ELISA) methods. In particular, measuring IgM antibodies using the ELISA method is often the initial form of detection, which, if present, implies a recent or ongoing infection. The diseases for which a diagnosis based on IgM antibody reaction is useful include hepatitis A, EBV infection, rubella, measles, CMV infection, and, in part, arbovirus infection (3234353637). This method, especially ELISA, can be used to analyze many clinical samples at the same time. However, a specialist is needed to confirm the results and the detection of IgG requires samples taken at two times points. Furthermore, the sensitivity and specificity of this method are relatively low compared with real-time PCR. Therefore, although this technique remains one of the most-used methods in the clinical field, it needs improvement.
1.3 Virus antigen detection
Three standard methods are used to detect viral antigens directly; sandwich ELISA, and immunofluorescence and immunoperoxidase assays.
Performing sandwich ELISA involves at least two antibodies that can specifically and independently bind to a viral antigen for detection, as two specific antibodies have a different binding domain for the viral antigen, to escape the interference phenomenon (38). Examples of the use of viral antigens in a clinical setting include the detection of respiratory viruses in pharyngeal inhalation fluids, such as the respiratory syncytial virus, influenza A and B types; the detection of enteric viruses in stool, such as adenovirus and rotavirus; the detection of the CMV, the herpes simplex virus, and the VZV in skin scrapes; the detection of the hepatitis B and C viruses in serum (39404142). The main advantage of this method is that it can be performed quickly (the results can be obtained within hours). Technologically, however, this method is often tedious and time consuming, difficult to read and interpret, and less sensitive and specific. Moreover, the quality of the test specimen obtained is extremely important for the proper running of this test.
The immunofluorescence method is mainly used for microbiological samples. It is based on fluorescence microscopy and is defined as a procedure for detecting an antigen in a cell using an antibody. This technique can visualize the distribution of target molecules throughout the sample, because the fluorescent dye is bound to specific biomolecules within the cell. However, a specialist is required to distinguish between false- and real-positive results, and it is difficult to apply this method to the mass diagnosis of many clinical samples at the same time.
Immunoperoxidase is a type of immunostain that is used in molecular biology, medical research, and clinical diagnostics. In particular, immunoperoxidase reactions include immunohistochemical or immunocytochemical procedures in which the antibodies are visualized via a peroxidase-catalyzed reaction. This method has been generally used to stain viral antigens or specific targets in clinical pathological tissues. However, it is also difficult to apply it to mass diagnosis.
1.4 Viral nucleic acid detection
Nucleic acid testing is a molecular technology that detects viral DNA or RNA. The detection of specific DNA and RNA from viruses is mainly performed by PCR. These molecular techniques are commonly used to obtain positive serological results because of their high sensitivity and specificity. Modifications of PCR, such as real-time PCR, can be used to determine viral load in addition to virus detection in sera from patients. For example, modified PCR methods are often used to monitor treatment success in patients with HIV. In some cases, the PCR product is sequenced to predict/confirm the genotype of the virus. The identification of the genotype of viruses is important because different genotypes can lead to different transmission pathways, virulence, and treatment. In some cases, certain mutated genes in patients are tested to determine the susceptibility to antiviral therapy and infection (252627). The use of this method has increased dramatically in recent years in the field of viral diagnostics because of improved efficiency and accuracy, the convenience of automation, and its cost effectiveness.
2. Method for the purification of viral nucleic acids
Viral diagnostics in a clinical setting directly affects patient treatment and outcomes. Furthermore, it provides important information regarding treatment decisions, hospital infection control, length of patient stay, hospital and laboratory costs, and laboratory efficiency (43). Therefore, accurate and rapid diagnosis is necessary for patient recovery. To this end, proper selection, collection, transportation, and storage of patient specimens must be performed correctly, to ensure accurate virus diagnosis (43). The WHO and the CDC are constantly updating the collection and specimen management of viral diseases. For example, the CDC reports collection protocols, storage procedures, and recommended clinical specimens for respiratory diseases (https://www.cdc.gov/urdo/downloads/speccollectionguidelines.pdf). In fact, the methods used for sampling, storage, and delivery affect the diagnostic results. Among them, the isolation of nucleic acids from viruses is the most important step in PCR and real-time PCR. This procedure can be largely classified into five methods that use boiling, an ion-exchange matrix, DNA precipitation, magnetic glass particles, and a silica membrane to isolate and purify the viral genome, respectively (4445). Viral nucleic acid extraction should be performed according to the amount of the specimen and the type of specimen and virus, and pure nucleic acids can be used for PCR. In the case of pure nucleic acids, the 280/260 ratio and the 280/230 ratio are close to 2, which is an index that can be used to increase PCR efficiency (http://www.nhm.ac.uk/content/dam/nhmwww/our-science/dpts-facilities-staff/Coreresearchlabs/nanodrop.pdf).
3. Application of molecular diagnostic techniques in clinical virology
Molecular diagnostics is a collection of techniques that are used to analyze biological markers in genomes and proteomes by applying molecular biology to medical tests. This technology is used to diagnose and monitor disease, to detect risks, and to determine the best treatment for individual patients (46). Molecular techniques include quantitative PCR (real-time PCR), multiplex PCR, DNA microarray, in situ hybridization, DNA sequencing, antibody-based immunofluorescence analysis, molecular profiling of pathogens, and bacterial gene analysis for antimicrobial resistance (21).
Real-time PCR is a standard molecular biology laboratory technique based on PCR. Real-time PCR is one of the fastest and most sensitive detection methods and has the advantage of being able to measure quantitative values. Two common methods for detecting viral DNA or RNA in real-time PCR use nonspecific fluorescent insertion dyes with any double-stranded DNA or oligonucleotide sequence-specific probes labeled with a fluorophore. Between these two methods, a sequence-specific probe relies on a DNA-based probe with a fluorescent reporter at one end and a fluorescence extinction at the other end. As a diagnostic tool, probe-based real-time PCR can be used for the quick detection of viral nucleic acids that cause infectious diseases. The introduction of probe-based real-time PCR assays in clinical microbiology laboratories has greatly improved the diagnosis of infectious diseases, and it is currently used as a tool for detecting emerging diseases, such as new influenza viruses, in diagnostic tests (224748).
4. Application of probe-based real-time PCR
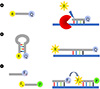
The probe-based real-time PCR allows sensitive and specific detection using fluorescent probe technology. In clinical trials, three types of nucleic acid detection methods are used most often, together with a real-time PCR test platform: hydrolysis probe (or TaqMan probe), molecular beacon, and hybridization probe (Fig. 1). First, the hydrolysis probe format uses oligonucleotides with a fluorescent label (reporter dye) at the 5′ end and another fluorescent label (quencher dye) at the 3′ end. The hydrolysis probe first binds to the target DNA, and then the 5′ exonuclease activity of the polymerase cleaves the probe during the primer extension step of the PCR process. Fluorescence emission can be detected from the reporter dye that is separated from the quencher dye (49). Second, the molecular beacon format is a molecule in the form of a hairpin that has an internally quenched fluorophore. When the probe sequence is hybridized to its target, the quencher dye is separated from the reporter dye and fluorescence is restored (50). Finally, the hybridization probe format uses two different fluorescence-labeled oligonucleotides. The donor probe has a fluorescein label at the 3′ end, while the receptor probe is labeled with a different fluorescein label at the 5′ end. When fluorescein in the donor probe is excited, it emits fluorescence at a specific wavelength. The sequences of the two probes are selected to hybridize to the amplified DNA fragment in a head-to-tail arrangement, to bring the two fluorescent dyes near to one another; the emitted energy leads to the exit of the dye attached to the acceptor probe and the emission of fluorescence at longer wavelengths. The increase in the amount of fluorescence measured is proportional to the increase in the amount of DNA generated during the PCR process. As the signal is emitted when both oligonucleotides are hybridized, fluorescence is measured immediately after the annealing step. After annealing, the temperature is raised and the hybridization probe is displaced by the polymerase. At the end of the elongation step, the amplification product is double stranded and the probe is too far away to allow fluorescence resonance energy transfer (FRET) (51). Moreover, real-time PCR techniques have been researched as a new method for multiplexing by melting curve analysis using probes such as peptide nucleic acids or locked nucleic acids (LNAs) (525354).
All detection methods rely on FRET (55). Among these detection methods, the hydrolysis probe format is commonly used clinically, is relatively easy to design, and has high sensitivity and specificity (22). The hydrolysis probe is designed according to the following rules. First, the amplification length of real-time PCR should be as short as 50~150 base pair. Second, the ratio of the GC sequence of the primers should be within the range of 20% to 80%. Third, it is recommended that the melting points (Tm) of the primers should be similar to each other and have a specificity through the GC ratio that is as high as possible. Fourth, the hydrolysis probe should be placed as close to the forward primer as possible, without overlapping. Fifth, as guanine (G) has the effect of limiting fluorescence expression, the hydrolysis probe should not contain a G at the 5′ end and the number of Gs should be lower than the number of cytosines (C). Sixth, the melting point of the TaqMan probe should be 8~10℃ above the melting point of the primer (https://eu.idtdna.com/pages/decoded/decoded-articles/pipettips/decoded/2013/10/21/designing-pcr-primers-and-probes) (56).
When designing probes and primers initially for real-time PCR technology, the following factors must be considered:
Nn = N0 × (1 + E)n,
the difference between the database access number of each target gene and the reference gene, the exon location of each primer and all probes, and the sequence of other viruses or types. The specific primers and probes for the target gene of the virus should collect as much information as possible regarding the virus strain and should be designed by selecting a conserved region (57). In silico tools such as BLAST or equivalent specificity are useful for analytical design. However, specificity must be empirically verified by direct experimental evidence from gel electrophoresis, melting profile, DNA sequencing, amplicon size, and/or restriction enzyme digestion. The algorithm for predicting the Tm of oligonucleotides is useful for their initial design, but the actual optimum annealing temperature must be determined experimentally. Although optimization of the primer does not follow a trend, it is clear that poor thermal optimization has a large impact on the quality of the analysis. Ideally, some evidence of primer optimization should be presented in the form of annealing temperature or Mg2+ details (58). Researchers should also check the tools used and record all PCR cycling conditions. Because the consumables that are used affect thermal cycling, the use of a single tube, strip, or plate, and its manufacturer must be identified. The level of transparency of plastic products (white or transparent) is also important because plastics exhibit substantial differences in fluorescence reflection and sensitivity (59).
In addition, real-time PCR using hydrolysis probes requires very careful analysis. More generally, the amplification reaction follows this equation: where Nn is the number of PCR amplifications after n cycles, N0 is the initial number of template replicates in the sample, E is the PCR efficiency, and n is the number of cycles (60). This equation is based on the assumption that there is initially one template in the reaction and the PCR efficiency is 100%. In other words, as the PCR efficiency is 100%, accurate data can be obtained (60). To do so, the standard control used to create the standard curve in real-time PCR must be accurate. A threshold Cycle value of 3.32 is best when 10-fold dilution of the standard control at high concentration is possible (60).
Moreover, multiplex real-time PCR should be different from the single channel, to check for inter-channel interference. The real-time PCR instrument should have a limited number of channels, to limit the number of multiplexed targets. Different channels may interfere with each other, depending on the Taq polymerase and probe. Multiplexing is still limited to four target/reporter dyes. Competition for a PCR master mix between two or more components in a multiplex analysis of a limited number of detector channels is a commonly used PCR platform. In addition, the design of all primer pairs should be optimized, so that all primer pairs can operate at the same annealing temperature during PCR (6162).
The real-time PCR reagents used in the clinical field may have technical deficiencies that affect analytical performance, depending on the developer's ability. Incorrect sample storage, poor preparation of nucleic acids, poor purity of the nucleic acids, and wrong selection of probes for PCR, and improper data and statistical analyses can lead to inaccurate results. Therefore, the real-time PCR reagents used in clinical trials are subjected to various performance evaluations during the development or quality control (QC) stage.
Among the validation guidelines of the real-time PCR method, the minimum information for publication of quantitative real-time PCR experiments (MIQE) guidelines are the most promising (63). The main issues in the performance evaluation of these guidelines among the performance evaluation items are analytical sensitivity, analytical specificity, accuracy, repeatability, and reproducibility (summarized in Table 2). First, analytical sensitivity refers to the minimum number of copies of a sample that can be accurately measured in the analysis. Analysis specificity refers to a real-time PCR analysis that detects the appropriate target sequence present in the sample over other nonspecific target sequences. Moreover, accuracy is the difference between the experimentally measured concentration and the actual concentration, and in the case of quantitative testing, it is shown as a copy. Repeatability indicates the accuracy and robustness of the analysis using the same sample repeatedly and analyzing it in the same way. Accuracy and repeatability can use the number of copies or standard deviation (SD) and coefficient of variation (CV) values (64). Reproducibility refers to differences in results between runs or between laboratories, and is typically expressed in SD or CV of copy numbers or concentrations (65).
DISCUSSION
Since the initial description of the PCR method, PCR-based techniques have been used clinically and scientifically to detect pathogens qualitatively or quantitatively, to identify mutations in pathogen genes, or to identify the types of pathogens (66676869). PCR-based techniques include restriction fragment length polymorphism, quantitative real-time PCR, sequencing, and modified PCR methods such as mutant enrichment with 3′-modified oligonucleotides (MEMO), as well as classic PCR (70717273).
Among these techniques, real-time PCR has revolutionized the diagnosis of human pathogenic viruses in clinical laboratories (74). Thus, real-time PCR plays an important role in the detection, quantification, and typing of viral pathogens in diagnostic methods related to viruses. In particular, real-time PCR with reverse transcription for the diagnosis of RNA viruses reduces the number of cDNA production steps and provides appropriate sensitivity and specificity within a short period (75). However, the limitations of real-time PCR include a difficulty in distinguishing live from dead cells, and it requires the identification in bacterial strains of phenotypic and biochemical characteristics (76).
Real-time PCR represented the introduction of the concept of real-time monitoring of DNA amplification through fluorescence monitoring (49). The fluorescence of the real-time PCR is measured at each cycle and the intensity of the fluorescence signal reflects the instantaneous extent of DNA amplification. In principle, the absolute amount of sample DNA can be determined according to a calibration curve that is proportional to the initial number of template DNA molecules in the sample using a point at which the fluorescence intensity increases above a detectable level (7778). Nonetheless, real-time PCR may be subjective, depending on the manner in which it is analyzed. Non-standard real-time PCR analyses can produce false-positive and falsenegative results based on the height of the threshold and the slope of the amplification curve. Thus, standard documentation and procedures are needed to determine the basic functional parameters of real-time PCR in the field of routine microbiological diagnosis (60).
The MIQE guidelines include instructions that provide the minimum information necessary to evaluate the real-time PCR experiment (63). In addition, intra-laboratory and inter-laboratory accuracy is often monitored by external QC programs. Additional clinical laboratory requirements include criteria for generating reporting results, whether samples are repeatedly measured, data from a false-positive/false-negative data analysis, and similarity between results from multiple laboratories using the same or different technologies (79). According to the MIQE guidelines and various performance evaluation guidelines (6080), the developed real-time PCR method should be validated based on analysis performance and clinical trial evaluation.
In conclusion, real-time PCR technology offers several important benefits. Simultaneous amplification and quantification requires additional manipulation of the PCR product, which reduces the risk of contamination and allows rapid processing of a large number of specimens. After sufficient validation, the standardized real-time PCR method is sensitive, has a wide detection range, and allows a quick and easy diagnosis.


 XML Download
XML Download