

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abstract
Deoxyribonucleotides (dNTPs) are important for the efficient growth of DNA viruses. Therefore, many DNA viruses have strategies for the upregulation of cellular dNTP levels. Both α- and γ-herpesviruses encode functional homologs of cellular dNTP anabolic enzymes, including the class I ribonucleotide reductase (RNR) large (R1) and small (R2) subunits, whereas β-herpesviruses modulate host cells to induce genes that increase dNTP levels. Interestingly, β-herpesviruses still express the nonfunctional RNR R1 subunit. However, it is not clear why β-herpesviruses still carry inactive R1 homologs. Recently, the R1 homologs of herpesviruses have been shown to inhibit innate immune signaling pathways. In particular, both functional and nonfunctional R1 homologs target receptor-interacting protein kinase 1 (RIP1) and inhibit RIP1-mediated signaling pathways to promote viral replication. Here, we summarize recent findings on the activity of herpesviral R1 homologs and discuss their roles in the regulation of innate immune signaling pathways.
Go to : 
REFERENCES
1). Jordan A, Reichard P. Ribonucleotide reductases. Annu Rev Biochem. 1998; 67:71–98.

2). Lembo D, Brune W. Tinkering with a viral ribonucleotide reductase. Trends Biochem Sci. 2009; 34:25–32.
3). Nordlund P, Reichard P. Ribonucleotide reductases. Annu Rev Biochem. 2006; 75:681–706.
4). Jacobson JG, Leib DA, Goldstein DJ, Bogard CL, Schaffer PA, Weller SK, et al. A herpes simplex virus ribonucleotide reductase deletion mutant is defective for productive acute and reactivatable latent infections of mice and for replication in mouse cells. Virology. 1989; 173:276–83.
5). Idowu AD, Fraser-Smith EB, Poffenberger KL, Herman RC. Deletion of the herpes simplex virus type 1 ribonucleotide reductase gene alters virulence and latency in vivo. Antiviral Res. 1992; 17:145–56.
6). Heineman TC, Cohen JI. Deletion of the varicella-zoster virus large subunit of ribonucleotide reductase impairs growth of virus in vitro. J Virol. 1994; 68:3317–23.
7). Aurelian L. Herpes simplex virus type 2: unique biological properties include neoplastic potential mediated by the PK domain of the large subunit of ribonucleotide reductase. Front Biosci. 1998; 3:d237–49.
8). Brune W, Menard C, Heesemann J, Koszinowski UH. A ribonucleotide reductase homolog of cytomegalovirus and endothelial cell tropism. Science. 2001; 291:303–5.
9). Upton JW, Kaiser WJ, Mocarski ES. Cytomegalovirus M45 cell death suppression requires receptor-interacting protein (RIP) homotypic interaction motif (RHIM)-dependent interaction with RIP1. J Biol Chem. 2008; 283:16966–70.
10). Mack C, Sickmann A, Lembo D, Brune W. Inhibition of proinflammatory and innate immune signaling pathways by a cytomegalovirus RIP1-interacting protein. Proc Natl Acad Sci U S A. 2008; 105:3094–9.
11). Rebsamen M, Heinz LX, Meylan E, Michallet MC, Schroder K, Hofmann K, et al. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep. 2009; 10:916–22.
12). Fliss PM, Jowers TP, Brinkmann MM, Holstermann B, Mack C, Dickinson P, et al. Viral mediated redirection of NEMO/IKKgamma to autophagosomes curtails the inflammatory cascade. PLoS Pathog. 2012; 8:e1002517.
13). Krause E, de Graaf M, Fliss PM, Dolken L, Brune W. Murine cytomegalovirus virion-associated protein M45 mediates rapid NF-kappaB activation after infection. J Virol. 2014; 88:9963–75.
14). Upton JW, Kaiser WJ, Mocarski ES. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe. 2010; 7:302–13.
15). Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2012; 11:290–7.
16). Guo H, Omoto S, Harris PA, Finger JN, Bertin J, Gough PJ, et al. Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe. 2015; 17:243–51.
17). Dufour F, Sasseville AM, Chabaud S, Massie B, Siegel RM, Langelier Y. The ribonucleotide reductase R1 subunits of herpes simplex virus types 1 and 2 protect cells against TNFalpha- and FasL-induced apoptosis by interacting with caspase-8. Apoptosis. 2011; 16:256–71.
18). Patrone M, Percivalle E, Secchi M, Fiorina L, Pedrali-Noy G, Zoppe M, et al. The human cytomegalovirus UL45 gene product is a late, virion-associated protein and influences virus growth at low multiplicities of infection. J Gen Virol. 2003; 84:3359–70.
19). Phillips SL, Bresnahan WA. Identification of binary interactions between human cytomegalovirus virion proteins. J Virol. 2011; 85:440–7.
20). Kim YE, Oh SE, Kwon KM, Lee CH, Ahn JH. Involvement of the N-Terminal Deubiquitinating Protease Domain of Human Cytomegalovirus UL48 Tegument Protein in Autoubiquitination, Virion Stability, and Virus Entry. J Virol. 2016; 90:3229–42.
Go to :
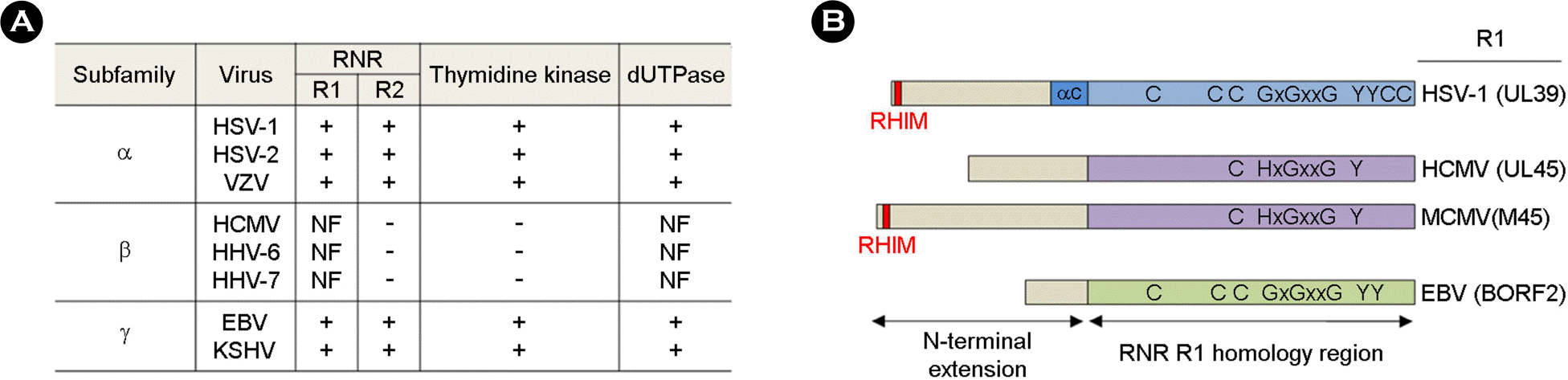 | Figure 1.Distribution and organization of herpesvirus R1 proteins. (A) Unlike human α- and γ-herpesviruses, human β-herpesviruses lack genes encoding the RNR R2 subunit and thymidine kinase and encode a nonfunctional RNR R1 subunit and dUTPase. (B) The C-terminal region of β-herpesvirus R1 proteins shares homology with other viral counterparts, but lacks most of the residues known to have a direct catalytic role. The proposed nucleotide-binding site (GxGxxG) and the catalytically active sites, i.e., five cysteines and two tyrosines (2), are shown. NF: nonfunctional. |
 XML Download
XML Download