

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abstract
Hepatitis A virus (HAV) positive stool samples were collected from acute hepatitis A patients during the two study periods of 2002 and 2011 in Seoul, South Korea, and their genetic variability was determined. From a total of 79 specimens, the nucleotide sequences of the VP1 and 2A junction were successfully amplified in 27 (34.2%) samples and subjected to sequence analysis. Genetically, there was a dramatic change in HAV subgenotypes from IA to IIIA during the past ten years. Sequence analysis identified that most strains belonged to genotype I, which is the main genotype globally. The subgenotype IA (93.3%, n=13/14) was the major subgenotype in 2002, whereas the subgenotype IIIA (69.2%, n=9/13) was predominant in 2011. Interestingly, a IIIA strain was identified from a patient who had a history of travel to India in 2002. The finding presented provides new insight into the genetic shift of circulating HAVs in South Korea.
Go to : 
REFERENCES
1). Bernard N, Fields DMK, Peter M. Howley. Fields Virology. 3 ed.Philadelphia: Lippincott-Raven;1996.
2). Pérez-Sautu U, Costafreda MI, Lite J, Sala R, Barrabeig I, Bosch A, et al. Molecular epidemiology of hepatitis A virus infections in Catalonia, Spain, 2005–2009: Circulation of newly emerging strains. J Clin Virol. 2011; 52:98–102.

3). Melnick JL. Properties and Classification of Hepatitis A Virus. Vaccine. 1992; 10:S24–6.
4). Jeon HB, Park JH, Lee HH, Kim DH, Lee HY, Shon DH, et al. Enhancement of Protein Productivity of Recombinant Hepatitis A Virus VP1 in Stably Transfected Drosophila melanogaster S2 Cells. J Bacteriol Virol. 2012; 42:69–75.
5). Robertson BH, Jansen RW, Khanna B, Totsuka A, Nainan OV, Siegl G, et al. Genetic Relatedness of Hepatitis A Virus Strains Recovered from Different Geographical Regions. J Gen Virol. 1992; 73:1365–77.
6). Lemon SM, Robertson BH. Current Perspectives in the Virology and Molecular Biology of Hepatitis A Virus. Semin Virol. 1993; 4:285–95.
7). Song YB, Lee JH, Choi MS, Koh KC, Paik SW, Yoo BC, et al. The age-specific seroprevalence of hepatitis A virus antibody in Korea. Korean J Hepatol. 2007; 13:27–33.
8). Yun H, Lee HJ, Jang JH, Kim JS, Lee SH, Kim JW, et al. Hepatitis A Virus Genotype and Its Correlation With the Clinical Outcome of Acute Hepatitis A in Korea: 2006–2008. J Med Virol. 2011; 83:2073–81.
9). Song HU, Hwang SG, Kwon CI, Lee JE, Ko KH, Hong SP, et al. Molecular Epidemiology of Hepatitis A Virus in the South-East Area of Gyeonggi-do in Korea. Yonsei Med J. 2009; 50:512–6.
10). Sohn YM, Rho HO, Park MS, Park JH, Choi BY, Ki M, et al. The changing epidemiology of hepatitis A in children and the consideration of active immunization in Korea. Yonsei Med J. 2000; 41:34–9.
11). Lee H, Cho HK, Kim JH, Kim KH. Seroepidemiology of Hepatitis A in Korea: Changes over the Past 30 Years. J Korean Med Sci. 2011; 26:791–6.
12). Mohd Hanafiah K, Jacobsen KH, Wiersma ST. Challenges to mapping the health risk of hepatitis A virus infection. Int J Health Geogr. 2011; 10:57.
13). Byun KS, Kim JH, Song KJ, Baek LJ, Song JW, Park SH, et al. Molecular epidemiology of hepatitis A virus in Korea. J Gastroenterol Hepatol. 2001; 16:519–24.
14). Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000; 132:365–86.
15). Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997; 25:4876–82.
16). Felsenstein J. PHYLIP (Phylogenetic Inference Package). 3.5c ed.University of Washington: Department of Genetics;1993.
17). Jukes TH, Cantor CR. Evolution of protein molecules. HN M, editor. editor.New York: Academic Press;1969.
18). Kwon OS, Byun KS, Yeon JE, Park SH, Kim JS, Kim JH, et al. Detection of hepatitis A viral RNA in sera of patients with acute hepatitis A. J Gastroenterol Hepatol. 2000; 15:1043–7.
19). Khanna B, Spelbring JE, Innis BL, Robertson BH. Characterization of a Genetic Variant of Human Hepatitis A Virus. J Med Virol. 1992; 36:118–24.
20). Lee D, Cho YA, Park Y, Hwang JH, Kim JW, Kim NY, et al. Hepatitis A in Korea: Epidemiological shift and call for vaccine strategy. Intervirology. 2008; 51:70–4.
21). Jeong SH, Lee HS. Hepatitis A: Clinical Manifestations and Management. Intervirology. 2010; 53:15–9.
22). Van Herck K, Jacquet JM, Van Damme P. Antibody Persistence and Immune Memory in Healthy Adults Following Vaccination With a Two-Dose Inactivated Hepatitis A Vaccine: Long-Term Follow-Up at 15 Years. J Med Virol. 2011; 83:1885–91.
23). Yoon YK, Yeon JE, Kim JH, Sim HS, Kim JY, Park DW, et al. Comparative Analysis of Disease Severity Between Genotypes IA and IIIA of Hepatitis A Virus. J Med Virol. 2011; 83:1308–14.
Go to :
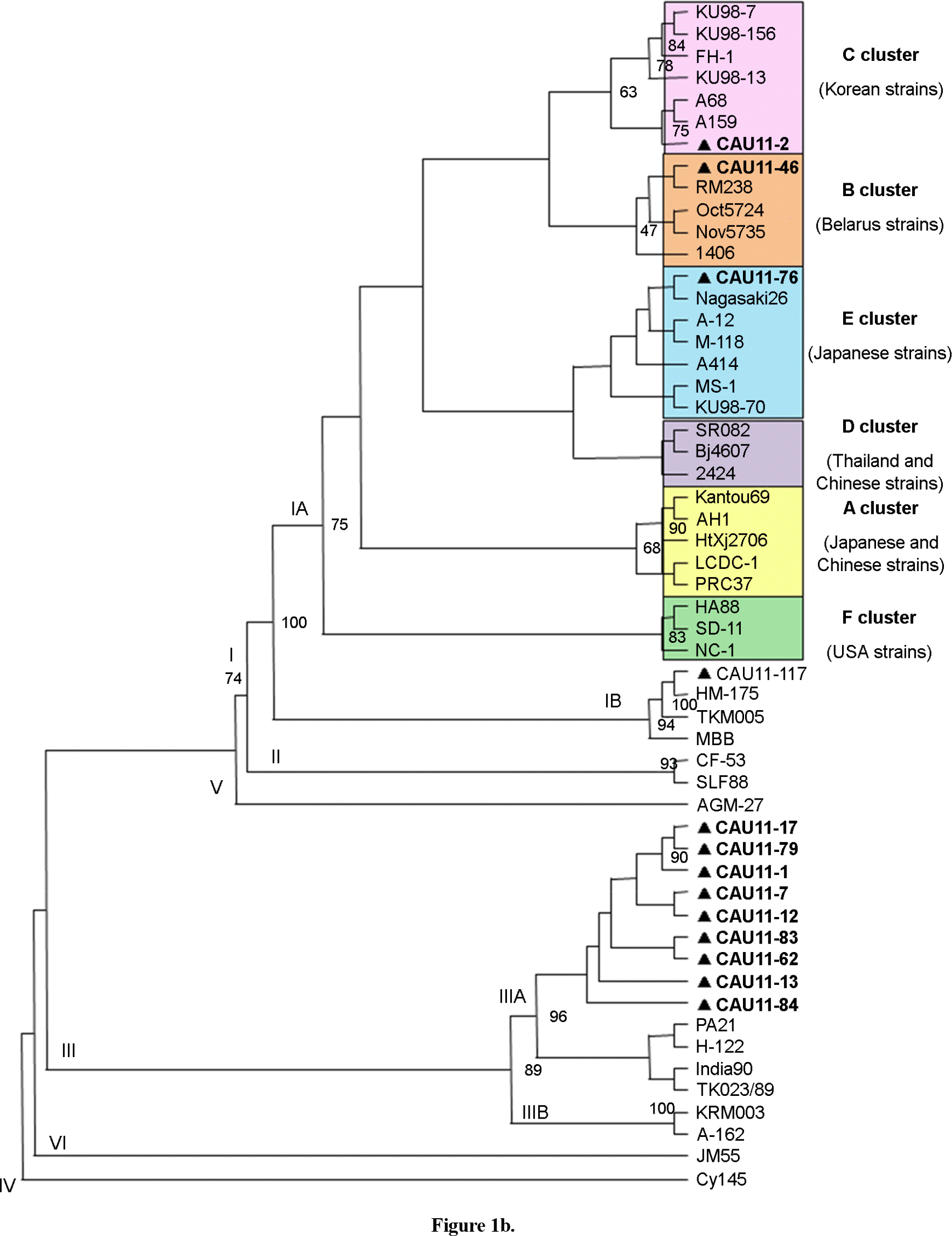 | Figure 1.Phylogenetic trees based on 168-bp HAV VP1/2A junction region nucleotide sequences show genetic relationships between 4 Korean isolates and 37 HAV strains obtained from the GenBank database. The shaded boxes indicate the 16 genotype I CAU isolates and the unfilled boxes indicate geographically related clusters. The numbers at the nodes indicate the level of bootstrap support (%) based on neighbor-joining analysis of 1,000 re-sampled datasets; only values above 50% are given. Hepatitis A virus Cy145 (L07732) was used as the out-group. (a) 2002 data; (b) 2011 data |
 XML Download
XML Download