

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Introduction
RNA polymerase, being at the focal point of gene regulation, became the most obvious target of choice in order to contain Mycobacterium tuberculosis infection. Success of the rifamycin group of antituberculosis agents-noteworthy being rifampicin, rifapentine, and rifabutin- was attributed to their capability to bind to and inhibit M. tuberculosis RNA polymerase (1). Over the years, Escherichia coli has served as the vantage point to comprehend the mechanisms of action of rifamycin and the resistance against them, thereof. During the same era, in the attempts towards eradication of tuberculosis, the world witnessed the fall and resurgence of this disease, especially in its dormant form. The failure of rifampicin as a first-line drug in the treatment of the latent form of tuberculosis triggered renewed attempts towards understanding the mechanisms that led to decreased efficacy of rifampicin in mycobacteria. The research that ensued opened up a Pandora's box, revealing mechanisms within mycobacterial species which ranged from ribosylative inactivation of rifampicin (2) to the sustained transcription activity induced by rifampicin itself (3). The latter led to the recognition of the interplay caused by protein-protein interactions between core RNA polymerase and sigma factors in contributing to the rifampicin resistome in mycobacteria (4). The discovery of RNA polymerase binding protein (RbpA) in S. coelicolor (5) and DnaA (replication initiation factor which promotes the unwinding or denaturation of DNA at oriC) in E. coli (6) paved the way for a new subset of such protein-protein interactions with prokaryotic RNA polymerase having implications on mitigation of inhibition by rifampicin. The exclusive occurrence of RbpA in a highly conserved form in actinobacteria, especially mycobacteria, has shed light over a possible lacuna that might have been overlooked ever since rifampicin was discovered and introduced as a drug.
We found support in the opinion expressed by Martinez (2008) (7), where he observed that a bulk of the antibiotics currently used for chemotherapy of infectious diseases have an environmental origin. The approach adopted in conducting studies towards understanding the role of antibiotics has been habitually confined to a clinical backdrop. Owing to the relatively low attention given to the role played by antibiotics in their original (non-clinical) environments, we perhaps have not noticed the clues that could have helped us predict and counter the emergence and evolution of antibiotic resistance. However, a discussion on the different mechanisms of rifampicin resistance will not be the major focus of this review. Instead we will try to view rifampicin as a small molecule which has an environmental origin. Ever since the discovery of rifamycin, there exists considerable amount of literature which treats it as an antibiotic. This approach has skewed the investigations towards drug research and has eventually resulted in a near elimination of attempts towards the discovery of novel roles of rifamycin in the context of microbial physiology. In this review, we attempt to comprehend the physiological response to rifampicin across the microbial spectrum. We will present the variation in this response among microbes and the implications of this interaction between rifampicin and the prokaryotic cell.
Major mechanisms of resistance to rifampicin
Rifamycin belongs to the class of antibiotics called ansamycins, which were initially discovered in a strain of Amycolatopsis mediterrenei (previously known as Nocardia mediterrenei) (8). This family of antibiotics is characterized by a planar naphthoquinone ring. Two positions have been extensively modified by hemisynthesis to improve the pharmacological properties and to yield commercial antibiotics such as rifampicin. Biochemical studies on the mechanism of inhibition of transcription by rifampicin have revealed that it blocks the initiation of transcription by preventing the synthesis of RNAs larger than dinucleotides. At the same time, rifampicin has no effect on the formation of the first phosphodiester bond and does not inhibit RNA elongation. These facts led to the proposal that rifampicin sterically blocks the path of nascent RNA during initiation (9~12). More than two decades later, the analysis of the three-dimensional structure of Thermus aquaticus RNAP in a complex with rifampicin revealed that the antibiotic binds near the RNAP active site at a protein pocket formed by the β-subunit (13). Rifampicin overlaps with the position of the third RNA nucleotide in the elongation complex (14). These data strongly supported the initial hypothesis on the steric mechanism of rifampicin action. From this structure, it was clear that the binding of such a large molecule primarily involves 12 amino acid residues. Since all of the rifampicin-resistant mutants have been mapped to the rpoB gene, rifampicin interacts only with the β-subunit encoded by rpoB, and mutation in any of these amino acids spontaneously generates a rifampicin-resistant phenotype. These mutations are clustered in a conserved region in the middle of the β-subunit.
Variation in the tolerance to rifampicin across the microbial spectrum
The minimum inhibitory concentration (MIC) for a drug/inhibitor has served as a universal indicator for monitoring the activity of antimicrobial agents. It is defined as the lowest concentration of an antimicrobial that will inhibit the visible growth of a microorganism after overnight incubation. Clinically, MIC values have not only served to determine the amount of antibiotic that the patient will receive but also the type of antibiotic used. Rifampicin has a broad spectrum of antibacterial activity. In 1983, Thornsberry and co-workers (15) conducted a comprehensive study on the spectrum of antibacterial activity of rifampicin. Their studies revealed that staphylococci were most susceptible (MIC 0.015 µg/ml), but most streptococcal strains had MICs ~ 1 µg/ml. Other susceptible species like Haemophilus influenzae, Neisseria gonorrhoeae, N. meningitidis, and Listeria monocytogenes had MICs of 1.0, 0.25, 0.03, and 0.12 µg/ml, respectively. The fast growing species of mycobacteria, Mycobacterium chelonei and M. fortuitum, were resistant, with MIC of >64 µg/ml. The MIC of M. smegmatis was ~20 µg/ml. However, the pathogenic species of mycobacteria, M. tuberculosis, has a MIC of 0.1~0.2 µg/ml (16). This sample survey clearly showed that response of microorganisms to rifampicin varies from a state of extreme sensitivity [as in the case of Staphylococcus pyogenes (MIC 0.008 µg/ml)] to a state of extreme tolerance [as in the case of Nocardia asiatica (MIC 300 µg/ml)]. On segregation of the sample into sets of pathogens, opportunistic pathogens and soil inhabitants, we found that the pathogens were more susceptible to rifampicin (MIC of rifampicin 0.008 to 8 µg/ml, shown in Fig. 1A), than opportunistic pathogens (MIC of rifampicin 1 to 64 µg/ml, shown in Fig. 1B). The soil microorganisms appeared recalcitrant to rifampicin (MIC of rifampicin 10 to >300 µg/ml, as shown in Fig. 1C).
The variation in the tolerance to rifampicin across microbial species has been attributed to modification in permeability (17, 18), efflux of rifampicin (19), modification of rifampicin (20~22) and duplication of rpoB (23, 24). The studies carried out on these aspects have been enlisted in Table 1. Apart from the above mentioned mechanisms, there have been some interesting observations of late, which have highlighted the novel concept of protein multifunctionality (25) caused by higher levels of proteome organisation in prokaryotes. On similar lines, it has been observed that some RNA polymerase-binding proteins (4, 5, 26~28) display a subsidiary function of reducing the susceptibility of RNA polymerase to rifampicin.
We studied the variation in the MIC values for rifampicin from available literature and classified the bacteria into Bacilli, Proteobacteria and Actinomycetales using the NCBI Taxonomy browser (www.ncbi.nlm.nih.gov/taxonomy). The 57 species of microbes were then grouped and their MIC values were plotted with respect to their groups (Fig. 2). Interestingly, we observed that there existed a class distinction among the microbes as far as their tolerance to rifampicin was concerned. The Bacilli have a MIC range from 0.008 µg/ml to 8 µg/ml. The Proteobacteria have a MIC range from 0.03 µg/ml to 64 µg/ml. The Actinomycetales show the highest tolerance levels to rifampicin with a MIC from 0.39 µg/ml to 300 µg/ml. Intrigued by this survey, we attempted to look into the probable reasons for increased levels of intrinsic resistance of Proteobacteria and Actinomycetales to rifampicin. As a representative of Proteobacteria we selected E. coli, on whom extensive studies have been carried out to determine antibiotic sensitivity profiles (29). The experiments carried out by Liu et al. (29) applied high-throughput screening of the Keio collection [an E. coli knockout collection of close to 4,000 strains, each with a different gene knocked out; Baba et al. (30)]. Kieo collection is a group of single-gene deletions of all nonessential genes in Escherichia coli K-12. It was constructed by replacing each ORF (open reading frame) with a kanamycin cassette. Out of the 4288 genes targeted, 3985 mutants were viable. This collection assists in the systematic analyses of unknown gene functions, gene regulatory networks and genome-wide scanning of mutational effects in a common strain background, E. coli K12 BW25113 (30).
Liu et al. (29) carried out experiments on the Kieo collection in order to look for mutants that were more susceptible to 1 of the 14 diverse antibiotics. Rifampicin was one of the antibiotics for which the susceptibility of the Kieo collection was also tested. The results revealed that there are genes, from different categories of bacterial function, which on being knocked-out resulted in increased vulnerability to rifampicin, although to differential extents (Table 2). This data provided a view of the complexity of intrinsic resistance to rifampicin in Proteobacteria.
Interference to the binding of rifampicin to RNA polymerase
We searched for similar kind of instances in Actinomycetales but could not come across any such study. However, there are individual reports from members of actinomycetes where it has been shown that inactivation of some genes have increased the susceptibility of organisms like M. smegmatis (2, 31) and S. coelicolor (5) to rifampicin (Table 3). Nevertheless, in a novel approach to comprehend the correlation between stationary phase induced changes in the mycobacterial polymerase assembly and phenotypic tolerance to rifampicin, we created a recombinant M. smegmatis mc2155 strain (SM07) which houses an affinity tagged RNA polymerase.
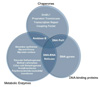
In this strain, the C-terminus of the β' subunit was fused to a hexa-histidine moiety (26). The simplified purification allowed delineation of the composition of the transcription complex at the stationary phase using 2D proteomics and tandem mass spectrometry. RNA polymerase purified from the mc2155 cells grown upto the late stationary phase was found to be rich with tightly associated proteins. When the 2D-PAGE profile of stationary phase RNA polymerase was compared with that of its exponential phase counterpart, it was observed that a substantial number of proteins in near or sub-stoichiometric amount remained bound to stationary phase RNA polymerase (26). The identities of these proteins were confirmed using MALDI-MS-MS sequence specific search (Table 4). When the stationary phase RNA polymerase was assayed against rifampicin for 50% inhibition of activity, it was found that its IC50 of rifampicin was twice that of exponential phase RNA polymerase (26). The assumption that formed the basis for testing rifampicin sensitivity was that any kind of physical interaction with another protein or a group of proteins (26), may reduce the accessibility of the enzyme to small molecules, which in this case was rifampicin. The identities of the associated proteins revealed broader classification into metabolic enzymes, chaperones and DNA-binding proteins (Fig. 3). The presence of many of these proteins in sub-stoichiometric amounts can be accredited to their association with different sub-populations of RNA polymerase during the growth cycle, where local milieus in the cytoplasm are fluctuating. Thus, it seems that all these proteins may shield RNAP during an assault of rifampicin. Proteins like chaperones and DNA-binding proteins are assigned the task of protection together with their probable reasons of being associated with any members of the transcription assembly. We tested the role of GroEL1 as a chaperone mediating the recruitment of other proteins to the stationary phase. For this purpose, RNA polymerase from ΔGroEL1 strain of M. smegmatis (32) was purified from the stationary phase. It was expected that the stationary phase enzyme from this mutant will show increased susceptibility to rifampicin. On the contrary, the enzyme was more resistant to rifampicin than a wild type RNA polymerase purified from stationary phase. Thus, the complex seemed to be more stable with recruitment of various proteins. Proteins like polyribonucleotide nucleotidyltransferase, 2-oxo acid dehydrogenases, acetyltransferase and DNA-RNA helicases form a part of the eubacterial mRNA degradosome, a multi component assembly (33). Pyruvate dehydrogenase in complex with biotinyl carboxylase was also found to be associated with the degradosome. Thus, we found that soil-dwelling actinomycetes like M. smegmatis have evolved higher levels of proteome organization so as to defend there transcription machinery from rifampicin that targets this essential component of their physiology. This could have serious implications on very high MIC values of rifampicin in these organisms. We came across a similar phenomenon from a comprehensive work carried out by Kuhner et al. (25) on Mycoplasma pneumoniae, where they attempted to comprehend the general rules of protein complex assembly and dynamics. Their work showed that heteromultimeric soluble protein complexes like RNA polymerase, ribosome, GroELs, and pyruvate dehydrogenase are into extensive sharing of components which eventually result in substantial crosstalk in the M. pneumonia protein complex network.
Metabolic enzymes get recruited to their sites of interaction on RNA polymerase to discharge their completely unrelated obligate functions (tolerance to rifampicin, in this case) and may dissociate to discharge their cellular activities under favorable conditions. In case of Providencia struartii, the enzyme 2'-N-acetyltransferase modifies bacterial peptidoglycan and the resemblance of its substrate with gentamycin enables it to transform both its normal substrate and the antibiotic (34). Thus, classification of a low specificity enzyme as a part of antibiotic resistance machinery adds on to the number of multiple resistance determinants in soil organisms (a general source of most natural antibiotics). However, similar roles of metabolic enzymes, associating with RNA polymerase, in modifying rifampicin remains to be proved.
Transcriptional response to rifampicin
The mechanism of action of rifampicin as an inhibitor of RNA polymerase activity has been a subject of active research. Several years of evidence have revealed that initiation of transcription is blocked by rifampicin, which binds to its site on the RNA exit channel and prevents the synthesis of RNAs larger than dinucleotides. Simultaneously, rifampicin does not influence the formation of the first phosphodiester bond and does not have an effect on RNA elongation. These facts led to the proposal that rifampicin sterically jams the corridor of nascent RNA during initiation (9, 11~13, 35). Biophysical studies on E. coli RNA polymerase (36) and the three dimensional structure of Thermus aquaticus RNA polymerase in complex with rifampicin (13) showed that the antibiotic binds near the RNA polymerase active site. The distance between rifampicin-binding site and RNA polymerase active site ranges between 18 to 20 Å (Å=Angstroms) (13, 36). There exists an overlap between rifampicin and the position of the third RNA nucleotide in the elongation complex (14). Thus, the steric model of rifampicin inhibition finds ample support from this data.
However, in a peculiar observation it was found that RNA polymerase from different bacteria elicit differential sensitivity towards rifampicin. Harshey and Ramakrishnan (37) showed that M. tuberculosis RNA polymerase is 1000 times more sensitive to rifampicin than its counterpart in E. coli. Contemporarily, Fabry et al. (38) showed that RNA polymerase from T. aquaticus was 100 times less sensitive to rifampicin as compared to E. coli RNA polymerase. In an innovative approach to gain insights into this riddle, Zenkin et al. (39) created a chimeric E. coli RNA polymerase in which the β-subunit segment encompassing rifampicin binding regions I and II (12) was swapped with the corresponding region from M. tuberculosis. However, this exercise did not confer any increased sensitivity to the chimeric E. coli RNA polymerase. But this result generated greater curiosity and led to the speculation that the differential sensitivities of RNA polymerases to rifampicin are not the sole prerogative of the regions which are involved in rifampicin binding, though it is subject to proof that structure of rifampicin binding pocket may be affected by differences in the nearby protein regions. Our experiments with rifampicin resistant RNA polymerases from M. smegmatis (27, 28) have revealed that although the different resistant strains have MICs of <300 µg/ml (SM0747), <400 µg/ml (SM0734) and <500 µg/ml (SM0748), their RNA polymerases have IC50 of 4.4 mg/ml, 6.4 mg/ml and 6.2 mg/ml. They also possessed differential affinity towards rifampicin (28). The strain (SM0748) harboring highest resistance to rifampicin (MIC <500 µg/ml) possessed 1,000 times lower affinity to RifR RNA polymerase (IC50 6.2 mg/ml) when compared to RifS RNA polymerase, while on the other hand RifR RNA polymerases (IC50 4.2 mg/ml and 6.4 mg/ml) from the other two RifR strains [MIC <300 µg/ml (SM0747) and <400 µg/ml (SM0734)] had only 10 times lower affinity to rifampicin when compared to RifS RNA polymerase. Investigations are currently underway to address this ambiguity through characterization of the rpoB gene from the wild type and mutant strains.
Though in a different niche, but in a related context it can be said that nature has been generous enough to endow some soil-inhabiting actinomycetes with two RNA polymerase β-subunit genes (24, 40~42). For the first time the genome sequence of Nocardia farcinica IFM 10152 showed the natural merodiplody involving duplicated rpoB alleles (40). The rpoB and rpoB2 genes share 88.8% identity in their gene sequences. The same group later reported the contribution of rpoB2 RNA polymerase β-subunit gene to rifampicin resistance in Nocardia species (42). However, it is unrealistic to consider the existence of a second rpoB as nature's blessing. Studies conducted subsequently to comprehend the physiological significance of rpoB duplication revealed an interesting discovery in Nonomuraea sp. strain ATCC 39727. It was found that the two rpoB paralogs, rpoB(S) and rpoB(R), provide the organism with two functionally distinct and developmentally regulated RNA polymerases (24), wherein the expression of rpoB(R) in Nonomuraea sp. and Streptomyces lividans led to the circumvention of the tight regulation of antibiotic synthesis by allowing constitutive activation of the antibiotic biosynthetic pathways. Additionally, there exists consensus regarding another aspect of the physiological significance of duplicated rpoB. It has been shown that RifR mutations in rpoB produce ancillary effects. RNA polymerases that harbor RifR RpoB elicit an increase or decrease in the expression of genes controlled by a stringent promoter. The decrease in expression was seen in case of E. coli promoters P1 and P2 of the rpoD operon, probably due to a negative control by stringent response. On the other hand, an increase in expression of some genes, viz. amino acid biosynthetic operons, was due to the positive control by stringent response (43). Thus, the existence of duplicated rpoB gene could be a part of an elaborate strategy that reduces the side effects of rpoB mutation. But the existence of duplicated rpoB is not comprehensive across species. We have recently reported the role of MsRbpA (an RNA polymerase interacting protein) in conferring phenotypic tolerance to rifampicin (27). As part of this work, we attempted to study the effect of MsRbpA on RifR RNA polymerases at their IC50 levels of rifampicin. The results showed that though RifS RNA polymerase was rescued by MsRbpA from rifampicin-mediated inhibition, a similar rescue effect was not visible on RifR RNA polymerase. Although we reasoned this observation to be a redundancy of the role of MsRbpA in a RifR background, we were intrigued by the similar levels of affinity shared by RifS and RifR RNA polymerases towards MsRbpA (28). This might imply that the interaction of MsRbpA with RifR RNA polymerases could be mitigating the side-effects of rpoB mutants by stabilizing the initiation complexes at stringently controlled promoters. This will subsequently lead to increased expression of genes that are positively controlled by the stringent response by providing more RNA polymerase molecules to initiate transcription at these stringently controlled promoters.
Dealing with rifampicin as an antibiotic before becoming resistant to it
Rifampicin has been reported to be inducing transcription of various sets of genes in B. subtilis (44) and in persistent cell populations of M. tuberculosis (3) at subinhibitory concentrations. Rifampicin induces δF in M. tuberculosis, whose homolog in B. subtilis is δB. Rifampicin was found to induce δB-dependent general stress response in B. subtilis (44). They showed that wild type B. subtilis survives better than sigB mutant on rifampicin-containing agar plates. Thus, rifampicin at subinhibitory concentrations acts as a causative agent of stress. The activation of ECF sigma factors is the characteristic of stress response in bacteria. Stress-induced mutagenesis is acknowledged as one of the ways in which non-growing cells temporarily increase their mutation rate to evolve into antibiotic-resistant variants (44). However, the mechanisms by which a RifS mother cell transforms into a RifR daughter cell/s has remained a mystery. Our work on MsRbpA has shown that it is effective in rescuing the transcription activity of a RifS RNA polymerase, but at the same time it was ineffective against RifR RNA polymerase (27). The in vivo levels of MsRbpA were highly sub-stoichiometric to the levels of RNA polymerase. These observations made it tempting to speculate about the protective role played by MsRbpA on a small fraction of RNA polymerase molecules when the remaining population of RNA polymerase has been left inactive due to the inhibitory action of rifampicin inside the cell. The physiological impact of this interaction between MsRbpA and RNA polymerase in the presence of rifampicin will be a small but essential level of transcription activity inside the cell. This activity may prove vital in the evolution of a RifR daughter cell/s (Fig. 4). As a proof of concept, a recent study revealed that asymmetry and aging in M. smegmatis lead to variable rifampicin susceptibility (45). The authors suggest that potential heritable factors influence a cell's tolerance to rifampicin and other drugs. Investigations are currently underway in order to ascertain this mechanism.
Unexplored facets of the character of rifampicin in nature
Subinhibitory concentrations of rifampicin (46) alter global transcription patterns (47). In this unconventional piece of work, the authors treated rifampicin as a small molecule that might act as a chemical signal in modulating metabolic processes at low concentrations. Their studies on E. coli and Salmonella typhimurium revealed that rifampicin can modulate transcription of a significant portion of genes. As far as the role of rifampicin as a small molecule in the chemical ecology of the environment is concerned, their data showed that it can be a significant component in the dynamics of bacterial communities in nature. This implies that inhibition occurs when high concentrations are attained and transcriptional modulation occurs at low concentrations. But going by the same fact, the situation becomes cumbersome and unexplored when we observe that soil bacteria can tolerate very high concentration of rifampicin as compared to pathogens and opportunistic pathogens. It will be interesting to find out the correlation between the differential level of tolerance and the variation in the role of rifampicin as a small molecule messenger. There have also been reports about the accumulation of rifampicin by microbial species like E. coli, S. aureus, M. tuberculosis, M. aurum and M. smegmatis (16, 48). The imbalance in the influx and efflux of rifampicin was cited as the reason for such kind of accumulation. But there could be further implications to such an accumulation of rifampicin, which stems from the recent finding where it was reported that many soil bacteria resorted to antibiotics as a means of nutritional source (49). Although the authors did not use rifampicin as one of the candidate antibiotics, it does not seem out of place to club the above two situations of accumulation of rifampicin and subsistence of soil microorganisms on antibiotics to regard rifampicin as a nutritional source for soil bacteria. This statement also gets corroborated by the existence of rifampicin modifying enzymes in nature (2). There might exist unexplored biochemical pathways which assimilate the end products of ribosylation, glycosylation, phosphorylation and decomposition of rifampicin. A focused study on how the rifamycin-producing organism Amycolatopsis mediterranei deals with rifamycin (precursor of rifampicin) will provide more clues into the realization of rifampicin as a nutritional source.
Conclusion
Over the past six decades, efforts of the scientific community have focused on the role of rifampicin as an inhibitor or a drug. However, in the last decade there have been novel findings as far as mechanisms of intrinsic tolerance to the drug is concerned. In spite of this, these findings have always been reported as advancements in the understanding of emergence of rifampicin resistance. E. coli has served as the model organism in most of these studies and indeed has been extremely prolific as a leading light towards the understanding of bacterial physiology. However, as a representative of soil or marine bacteria, or even commensal populations of living organisms, it has fallen short of expectations (50). The variation in the physiological response to rifampicin bears proof to the aforesaid scenario. One aspect of microbial physiology that has been highlighted by Davies (50) is the consideration of antibiotics as low molecular weight compounds. The world of these molecules-the Parvome- was predicted to be at least an order of magnitude larger than the number of prokaryotes in the biosphere. It will be interesting to decipher the role of rifampicin with respect to the parvome. Also, the fact that soil micro-organisms have been reported to be subsisting on antibiotics (49), makes it an interesting possibility to realize the metabolic pathway of assimilation of carbon and nitrogen from rifampicin.






 XML Download
XML Download