

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Haemophilus parasuis is a small, non-motile, pleomorphic gram-negative rod of the family Pasteurellaceae (1, 2). It is the causative agent of Glässer's disease, which is characterized by a fibrinous polyserositis, polyarthritis and meningitis. This illness is considered a sporadic, stress-associated disease of young pigs (2, 3).
Diagnosis of H. parasuis infection is generally made by isolation of the organism from a clinical specimen and subsequent biochemical analysis (2, 4, 5). However, recovery of H. parasuis from specimens is low, due in part to the fragility and fastidious growth requirements of H. parasuis relative to other bacteria that may also be present in the specimen (2, 6). In fact, a retrospective analysis conducted in diagnostic laboratories in Ontario, Canada indicated that the inability to confirm the presence of H. parasuis from submitted specimens (7) may have resulted in the true incidence of the disease being underestimated by as much as 10-fold. The polymerase chain reaction (PCR) provides fast and accurate identification of H. parasuis not only in pure culture but also in clinical samples, even when nonviable organisms are present (8). Recently, several PCR methods for H. parasuis have been developed (8, 9). Oliveira et al. (8) introduced a PCR method to detect 16S ribosomal DNA sequences. However, the 16S rDNA sequence is highly conserved and PCR primers designed to amplify the H. parasuis cognate may hybridize with 16S rDNA sequence of Pasteurellaceae. In the PCR, replicon with same size of H. parasuis was found for Actinobacillus indolicus (8). This makes the interpretation of a positive reaction difficult, especially when testing samples were obtained from the upper respiratory tract. However, recent report of Angen et al. (9) improved the weakness of 16S rDNA PCR of Oliveira et al. (8) by modifying primers for 16S rDNA region. In general, diverse PCR amplifying different regions of amplicon were developed in order to detect accurately even one pathogen such as Salmonella, Brucella and so on. Therefore, another PCR test based on specific gene sequences of H. parasuis is necessary to identify this porcine pathogen.
In this study, we cloned and sequenced a new gene of H. parasuis using a colony blot technique to determine an immunogenic antigen. Based on gene sequence comparison using GenBank data, the newly identified gene has 83% amino acid sequence similarity with an ABC transporter of H. ducreyi, which is an ATP-binding protein and permease. Since this species-specific gene was selected by comparative analysis among the gene from the bacteria within the Pasteurellaceae family, the PCR technique presented in this study could be a valuable method for diagnosis of H. parasuis infection from clinical materials.
MATERIALS AND METHODS
Bacterial strains and culture conditions
Fourteen reference strains for H. parasuis serovars 1 to 15, excluding serovar 8, were obtained from Dr. Chimmel in Germany (Bundesinstitut für gesundheitlichen Verbrauchershcutz und Veterinarmedizin, Fachberich Bakterielle Tierseuchen und Bekampfung von Zoonosen, J-Jena) and one H. parasuis standard strain (ATCC No. 19417) was purchased from American Tissue Culture Collection (ATCC). For propagation, all strains were inoculated on chocolate agar and incubated at 37℃ for 48 h. In this study, H. parasuis standard strain was used for genomic DNA extraction. All v-factor dependent strains were grown on chocolate agar, whereas all other strains were grown on sheep blood agar and nutrient agar. These cultures were incubated at 37℃ overnight.
Clinical samples
Seventy nine clinical samples from pigs suspected for being infected with H. parasuis, showing polyserositis or arthritis at necropsy were collected from 2007 to 2009 in Korea. Swabs were vortexed in 300 µl of PBS. DNA was extracted from the suspension using genomic DNA extraction kit (Intron Co., Sungnam, Korea).
Isolation of genomic DNA from H. parasuis
Genomic DNAs were obtained using DNAzol reagent (Invitrogen, Carlsbad, CA, USA). H. parasuis in culture medium [4 ml Tryptic soy broth with 5 µg/ml nicotinamide dinucleotide (NAD)] was centrifuged at 12,000 x g for 5 min. After centrifugation, the pellet was resuspended in 200 µl distilled water, and 1 ml of DNAzol reagent was added and mixed by gentle vortexing. The lysate was centrifuged at 10,000 x g for 10 min and the supernatant (1 ml) was transferred to a new tube. Genomic DNA from the supernatant was precipitated by adding 100% ethanol (500 µl) and washed with 75% ethanol. The dried DNA pellet was dissolved in 200 µl 0.1 N NaOH.
Library construction and screening
Standard molecular cloning techniques were used for library construction (10). Sau3A1 (Promega, Madison, WI, USA) fragments (0.5~3 kb) of partially digested H. parasuis genomic DNA were recovered from an agarose gel by using an Accuprep Agarose Gel Extraction Kit (Bioneer, Daejon, Korea) and cloned into the BamH1 site of pET-28a, pET-28b and pET-28c expression vectors containing the T7 promoter/lac operator and 6X His tag (Novagen, Madison, USA). For analysis of Escherichia coli transformants, the transformation mixture was plated on LB plates containing 50 µg/ml kanamycin and incubated overnight (16 h) at 37℃ until the colonies were 1~2 mm in diameter. The colonies were transferred to nitrocellulose membrane filters (BIO-RAD, Hercules, CA, USA). The identification of clones on colony blots was performed according to the manufacturer's protocol (The QIA expressionist, Qiagen, Valencia, CA, USA). After induction with 1 mM isopropyl-β-thiogalactopyranoside (IPTG; Promega) at 37℃ for 4 h, the colonies on the membranes were treated with denaturing solution (0.5 M NaOH, 1.5 M NaCl) and neutralization solution (1.5 M NaCl, 0.5 M Tris-HCl, pH 7.4). After washing with Tris-buffered saline (TBS: 10 mM Tris-HCl, pH 7.5, 150 mM NaCl) the membranes were blocked with blocking buffer [3% bovine serum albumin (BSA) in TBS] for 1 h. After washing twice in TTBS [100 mM Tris-HCl, 0.9% (w/v) NaCl, 0.1% (v/v) Tween 20, pH 7.5], for 10 min, the membrane was washed again in TBS for 10 min. The prepared membranes were reacted with an anti-6X His-tag mouse IgG monoclonal antibody (0.1 µg/ml; Novagen) and horseradish peroxidase (HRP)-conjugated anti-mouse IgG goat monoclonal antibody (Sigma, St. Louis, MO, USA). The HRP conjugate-bound membranes were developed with HRP staining solution (0.018% H2O2, 0.06% 4-chloro-1-naphthol, 20% methanol in 25 ml TBS), and the reaction was terminated by washing with tap water. The developed membranes were aligned with the original transformation plates and the anti-His tag positive clones were picked with sterile toothpicks and transferred into cluster tubes (1.2 ml, 96 wells, Costar 4411) containing LB broth with 50 µg/ml kanamycin. The cluster tube cultures were grown overnight at 37℃ and the sequences of the cloned genes were analyzed.
Sequence analysis of cloned genes
An ABI3730xl automatic sequencer (Applied Biosystems, Foster City, CA, USA) was used for analysis of the cloned inserts. SP6 promoter and T7 terminator primers were used as sequencing primers. DNA sequences were analyzed using the nucleotide BLAST algorithm [National Center for Biotechnology Information server (http://www.ncbi.nlm,nih.gov)] to identify homologous genes in the GenBank database.
PCR primers and PCR conditions
Based on the sequence data, a pair of primers specific for H. parasuis was designed: HP forward (5'- GAT CGA GTT GGA TGC CTG CAC GGT-3') and HP reverse (5'- GAT CCA TTG CAA ACC CTT CGC CCG-3'). The tubes containing bacteria were boiled for 10 min. After boiling, the tubes were centrifuged at 12,000 rpm for 5 min. The supernatant was used as the DNA template. A PCR was carried out in a 50 µl mixture containing 3 U Taq DNA polymerase (Promega), 3 mM MgCl2, 1X reaction buffer [5 mM Tris-HCl (pH 8.0), 10 mM NaCl, 0.01 mM EDTA, 0.1 mM DTT, 5% glycerol, 0.1% Triton X-100], 0.3 mM each dNTP (Promega) and 5 µl of extracted DNA. The PCR mixture was subjected to a 35-cycle PCR profile of 94℃ for 30 sec, 53℃ for 90 sec, 72℃ for 1 min, followed by a final extension step of 72℃ for 5 min. The PCR products were run in a 1% agarose gel for 50 min at 100 V. Gels were stained with 0.5 µg/ml ethidium bromide solution and photographed.
PCR sensitivity and specificity
Serial 10-fold dilutions of 24-h broth cultures containing 1.0 × 107 CFU/ml of H. parasuis (strain: ATCC 19417) were tested by PCR to determine the sensitivity of the proposed test. One milliliter of each dilution was used for DNA extraction. The tubes containing bacteria were boiled for 10 min. After boiling, the tubes were centrifuged at 12,000 rpm for 5 min. The supernatant was used as the DNA template and was also tested to determine the minimum amount of bacteria that could be detected in the samples. Specificity of the PCR test was determined by testing serovars of H. parasuis and 16 bacterial species commonly isolated from pigs (Table 1).
RESULTS
Isolation of H. parasuis polypeptide-expressing plasmid constructs
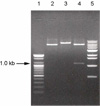
A total of 9 colonies out of about 2,500 showed a positive reaction against anti-His tag monoclonal antibody. Four colonies showing the strongest signals were picked. Plasmid DNA was extracted from the 4 colonies and the sequences of the inserts were analyzed. One of these clones, named 2A3, showed significant similarity to sequences in GenBank (see below) and was therefore chosen for further characterization. To verify the presence of an insert in the pET vector, clone 2A3 was digested with restriction enzymes (Fig. 1). Clone 2A3 digested with Not I (which cuts once in the vector) was linearized to a fragment larger than linearized pET vector (Fig. 1, lane 3), thus indicating the presence of an insert in 2A3. Because the H. parasuis genomic DNA was originally subjected to partial digestion with Sau3A1 and ligated into BamHI-digested vector, the 2A3 clone should have at least 2 Sau3A1 restriction enzyme sites (GATC), one or both (or neither) of which could also be part of a BamHI site (GGATCC). Digestion with BamHI released a fragment of about 1 kb from the vector (Fig. 1, lane 4), thus establishing the size of the insert.
Sequence of the 2A3 DNA insert
The 2A3 DNA insert-containing plasmid was subjected to further sequence analysis. Analysis of the determined DNA sequence (1,105 bp) revealed an open reading frame encompassing the entire DNA length but no putative gene in other frames. The sequence of the cloned DNA was submitted to GenBank (accession number DQ153243). BLAST analysis of the nucleotide sequence showed it to have 78% similarity with the ABC (ATP binding cassette) transporter/ATPase binding protein/permease gene of the H. ducreyi 35000HP strain. The size of H. ducreyi ABC transporter gene is 1,956 bp, and the cloned 2A3 sequence matched the H. ducreyi ABC transporter gene from nucleotide 142 to 1,247. Moreover, BLAST analysis showed 83% amino acid sequence similarity with that of H. ducreyi. Latest BLAST analysis showed this sequence showed following similarity with ABC transporter genes from some bacterial species; 95% similarity with H. parasuis, 76% similarity with Actinobacillus pleuropneumonia, and 74% similarity with H. somni.
PCR specificity and sensitivity
Two H. parasuis-specific primers were designed based on a species-specific gene and, as predicted, an H. parasuis-specific 1,105-bp PCR product was amplified from H. parasuis genomic DNA (Fig. 2). The PCR test detected 15 different H. parasuis serovars whereas the PCR result was negative for 16 other bacterial species that were tested (Table 1). The 2A3 PCR primer pair detected a minimum concentration of 1 × 104 CFU/ml of H. parasuis organisms (Fig. 3). The 2A3 DNA insert-specific bands were observed when the PCR was performed using DNA extracted from bacterial suspensions in the range of 107~104 CFU/ml. The bands at the bottom of the gel were observed in all samples, indicating unincorporated primers.
We also tested clinical samples from pigs suspected for being infected with H. parasuis. Of 79 samples tested, 30 samples (38.0%) were positive by PCR with 2ABC, while H. parasuis was recovered from only 8 (10.1%) samples, which all were positive by PCR test (Table 2).
DISCUSSION
Because of the fastidious growth requirements of H. parasuis, isolation and identification of this pathogen from clinical specimens have been challenging. To overcome these difficulties, several PCR methods were developed for rapid and accurate diagnosis of the bacterium (8, 11, 12). In addition, gene amplification by PCR can provide other important information, including data on pathogenicity or serovar, when the amplified PCR product is characterized by RFLP or sequence analysis (13). To diagnose H. parasuis infection by PCR, primers were designed based on the 16S small ribosomal DNA subunit of H. parasuis (8). However, the authors reported that their primers showed a weak false positive reaction against another member of the Pasteurellaceae family, Actinobacillus indolicus (8). Recently, the PCR using modified 16S rDNA primer reported by Angen et al. (9) improved the weakness of Oliveira's PCR (8) and the formal PCR was found to be complete species specific for H. parasuis. Oliveira reported a detection rate of 48.6% based on 16S ribosomal DNA PCR, and a bacterial isolation rate of 12.3% (14). The PCR detection rate and the bacterial isolation rate in our study were 38.0% and 10.1%, respectively. Although the samples tested were different in both studies, Oliveira's detection rate and isolation rate are 10.6% and 2.2% higher, respectively, than those reported in the present study. All samples testing positive in cultivation were also positive by PCR. However, 7 of 49 samples testing negative by PCR with 2A3 primers were positive by PCR with 16S rDNA primers (14). We cannot explain this difference in PCR results obtained with the two different primer pairs. The 16S rDNA sequences of A. pleuropneumoniae and H. parasuis have 89.8~90.9% similarity. Thus, some bacteria of this family may show false positive PCR results when H. parasuis 16S rDNA-based primers are used (9).
Genes encoding ABC transporter proteins are highly specific to the particular species (15). The ABC superfamily of active transporters is composed of about 50 functionally diverse prokaryotic and eukaryotic transmembrane proteins (16). These proteins transport a variety of substrates, including amino acids, lipids, inorganic ions, peptides, sugars, metals, drugs, and proteins (16). These proteins utilize energy derived from the hydrolysis of ATP to transport the substrate across the membrane against a concentration gradient (17). Because the amino acid sequence of the 2A3 polypeptide shows 83% similarity with that of an H. ducreyi ABC transporter, it is expected that the 2A3 protein also acts as a transporter. The similarity of nucleotide sequence between the 2A3 and the ABC transporter gene of H. ducreyi is 78%, and 99% identity with ABC type oligopeptide transport system in H. parasuis in recent BLAST analysis. However, the 2A3 sequence has 76% identity with A. pleuropnumoniae strains, which shows less similarity for 16S rDNA sequence. Therefore, PCR primers based on the putative ABC transporter gene 2A3 is thought to detect H. parasuis specifically. In a diagnostic real-time PCR for Mycoplasma hyopneumoniae, the agent of swine mycoplasma pneumonia, a pair of primers amplifying a putative ABC transporter gene was used (15). It showed high sensitivity and specificity. Likewise, the ABC transporter gene in this study could be a valuable PCR target for H. parasuis detection.
In conclusion, we analyzed a previously unknown gene encoding an ABC transporter-like protein of H. parasuis and developed a new PCR diagnostic tool using primers designed from the aforementioned sequence. The PCR product seems to be highly specific for H. parasuis, allowing the pathogen to be differentiated from other bacteria with appreciable sensitivity. Since this species-specific gene was selected by comparative analysis among the gene from the bacteria within the Pasteurellaceae family, the PCR technique could be a valuable method for diagnosis of H. parasuis infection from clinical materials.




 XML Download
XML Download