

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Metagenomics is the genomic analysis of microorganisms by direct extraction and cloning of DNA from their natural environment. For the purpose of this mini-review, the terms 'unculturable' and 'as yet uncultivated' are used to describe organisms that have yet not grown on artificial media in vitro. It is well established that only approximately 1% of bacteria on Earth can be readily cultivated in vitro - the so called 'great plate count anomaly' - based on the observation that microscopic counts are considerably larger than the equivalent total viable counts (1~3). There are currently estimated to be 61 distinct bacterial phyla, of which 31 have no cultivable representatives (4). The topology of the archaeal phylogenetic tree remains uncertain, but it is clear that the 54 species of Archaea cultured to date represent only a fraction of the total diversity, with 49 lineages mostly uncultured (5).
Because the majority of bacteria and Archaea remain unculturable, the diversity of complex bacterial communities is inevitably underestimated using standard cultivation methods. Furthermore, organisms of key importance to the community and the entire ecosystem or pathogens of plants and animals may be overlooked if they are unculturable. Consequently, with the development of molecular culture-independent techniques, there has been a move towards the characterization of mixed bacterial populations within biomass from the environment and in samples from animals (including humans) using PCR amplification of housekeeping genes, particularly the 16S rRNA gene, followed by cloning for purification and sequencing for identification (6, 7).
As a result, numerous novel phylotypes have been identified among bacterial communities isolated from a wide range of habitats: from seawater and soil to the health- and disease-associated microbiota of humans (8~10). Recently, 454 pyrosequencing has been used to investigate bacterial communities by sequencing 16S rRNA gene amplicons; examples of such studies include a study of nasopharyngeal microbiota in children (11), metagenomic analysis of the upper respiratory microbiota in pandemic influenza A (12), metagenomic analysis of human diarrhea (13), and analysis of the human distal gut microbiome (14, 15).
In the next section, I discuss Sanger-sequencing and next-generation DNA sequencing platforms. In addition, I highlight some applications of metagenomic techniques in the field of medical microbiology.
1. Sequencing Methods in Metagenomic Research
1-1. Sanger sequencing
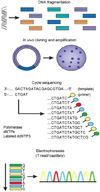
Since the early 1990s, DNA sequencing has been performed almost exclusively using capillary-based, semiautomated implementations of Sanger biochemistry (16~18) (Fig. 1). In high-throughput production pipelines, the DNA to be sequenced is prepared by one of two approaches. For shotgun de novo sequencing, randomly fragmented DNA is cloned into a high-copy-number plasmid, which is then transformed into Escherichia coli. Alternatively, for targeted resequencing, PCR amplification is carried out with primers that flank the target. The output of both approaches is an amplified template, either in the form of many 'clonal' copies of a single plasmid insert present within a spatially isolated bacterial colony that can be picked, or as many PCR amplicons present within a single reaction volume. The sequencing biochemistry takes place in a 'cycle sequencing' reaction, in which cycles of template denaturation, primer annealing, and primer extension are performed. The primer is complementary to the known sequence immediately flanking the region of interest. Each round of primer extension is stochastically terminated by the incorporation of fluorescently labeled di-deoxynucleotides (ddNTPs). In the resulting mixture of end-labeled extension products, the label on the terminating ddNTP of any given fragment corresponds to the nucleotide identity of its terminal position. Sequence is determined by high-resolution electrophoretic separation of the single-stranded, end-labeled extension products in a capillary-based polymer gel. The reported error rates of Sanger sequencing range from 0.001% (19, 20) to more than 1% (21, 22) and seem to depend on the software that is used for post-processing of the reads. Pyrosequencing, also known as "454 sequencing", produces shorter reads (23, 24). When the technology was first introduced, the read length was about 110 nt, but it has now increased to more than ~450 nt. Huse et al. (2007) reported an error rate of 0.49% for 100~200 nt-long reads (25), while the read simulation software MetaSim (22) produced reads with an error rate of 2.8% with parameters based on the original 454 publication (24). Pyrosequencing is still actively being researched and optimized (26).
1-2. Next-generation DNA sequencing
The current sequencing revolution is being driven by three commercially available platforms: the 454 (Roche), Genome Analyzer (Illumina/Solexa), and ABI-SOLiD (Applied Biosystems) (27, 28). These new technologies are based on different principles than the classical Sanger-based method (29), and these methods are collectively referred to as either 'next-generation' sequencing (NGS), 'high throughput' sequencing, 'ultra-deep' sequencing, or 'massively parallel' sequencing. The 454 system was the first NGS platform available as a commercial product (24) (Fig. 2). Using this approach, libraries can be constructed using any method that gives rise to a mixture of short and adaptor-flanked fragments. Clonal sequencing features are generated by emulsion PCR (30) with amplicons captured on the surface of 28 µm beads. During sequencing, one side of the semi-ordered array functions as a flow cell for introducing and removing sequencing reagents, whereas the other side is bonded to a fiber-optic bundle for charge coupled device (CCD)-based signal detection. During each of several hundred cycles, a single species of unlabeled nucleotide is introduced. On templates where this results in an incorporation event, a pyrophosphate is released. Due to the activity of ATP sulfurylase and luciferase, incorporation events immediately drive the generation of a burst of light, which is detected by the CCD as corresponding to the array coordinates of specific wells. In contrast with other platforms, therefore, this sequencing by synthesis must be monitored by the camera does not move relative to the array. Across multiple cycles, the pattern of detected incorporation events reveals the sequence of templates represented by individual beads. The sequencing is 'asynchronous' in that some features may get ahead or behind other features depending on their sequence relative to the order of base addition. A major limitation of the 454 technology is related to homopolymers. Because there is no terminating moiety that prevents multiple consecutive incorporations in a given cycle, the length of all homopolymers must be inferred from the signal intensity. In contrast, the Sanger method is based on DNA synthesis on a single-stranded template and di-deoxy chain termination (31, 32). Improvements in the cost-effectiveness of references and the development of high-throughput techniques have enabled direct sequencing of clones without laborious prior screening by restriction analysis (18). Relative to other next-generation platforms, the key advantage of the 454 platform is read length. For example, the 454 FLX instrument generates ~400,000 reads of 200 to 300 bp per instrument-run. Currently, the per-base cost of sequencing with the 454 platform is much greater than that of other platforms (e.g., SOLiD: Applied Biosystems and Solexa: Illumina) but it may be the method of choice for certain applications where long read lengths are critical (26).
2. Metagenomic Approaches For Discovering Novel Microorganisms
In this section, I highlight the utility of using metagenomic approaches to explore human microbial diversity and the potential diagnostic value of a metagenomic approach.
2-1. Determination of the diversity of bacterial communities (Bacteriome) using sanger sequencing technique
2-1-1. Background
Asthma is a heterogeneous airway inflammation syndrome with an increasing incidence in populations around the world. Its heterogeneity has made it difficult to determine pathogenesis and reasons for frequent exacerbation. Chronic infections cause a decline in lung function and may be a risk factor for the development and triggering of bronchial asthma. The association between respiratory infections and atopy or asthma remains to be determined and may be highly dependent on different infectious agents (33). The clinical relevance of the microbiota present on the oropharyngeal wall is unknown. However, it is known the diversity of the oral microflora can be altered by the use of antibiotics, probiotics, diet, and the microbiota in the gut. Changes in the microbiota of the GI tract can affect mucosal immunity in the lungs, as well as the gut, by bystander suppression (34). The link between oral and airway tolerance may be related to the induction or suppression of systemic immune tolerance due to challenge by antigens. Therefore, defining the characteristics of the oropharyngeal microbiota may increase our understanding of the pathogenic roles of these microorganisms and aid in the discovery of novel therapies for the prevention and treatment of oral complications (35). To define the difference in the oropharyngeal microbiota between patients with bronchial asthma and non-asthmatic controls, the 16S rRNA gene from bacterial species collected from healthy bronchial and asthma samples was analyzed by sequence analysis.
2-1-2. Comparison of bacterial microbiota between asthma patients and non-asthmatic controls
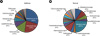
The diversity of the oropharyngeal swab sample microbiomes was characterized by sequencing the V3 region of the 16S rRNA gene that was amplified from genomic DNA samples of four asthma patients and four controls. About 50 clones were sequenced from each sample. Based on an analysis of 376 16S rRNA clones, the diversity of the microflora from the bronchial asthmatic and non-asthmatic control groups was striking; a total of 39 different bacterial genera representing six different bacterial phyla were documented in the non-asthmatic control samples. The majority of sequences in the metagenome library from the control group had Neisseria spp. (23.3%), Prevotella spp. (7.8%) and Streptococcus spp. (9.3%) according to GenBank, Eztaxon, and RDP database analysis tools. In contrast, a total of 21 different bacterial taxa were present in the samples from the group with bronchial asthma. The majority of sequences in the bronchial asthma library were identified as Streptococcus spp. (29.5%), Prevotella spp. (22.4%), Neisseria spp. (6.0%), and Veillonella spp. (13.7%). To examine the diversity of the microbial community present in oropharyngeal swab samples, assembled sequences were assigned to bacterial genera based on the taxonomy of their closest relatives as judged by Blastn analysis. While the two data sets showed broad taxonomic similarity, there was notable variation in each sample at the species level. It should be noted, however, that this analysis was coverage-limited. To correlate the variation in bacteria between swab samples, the Blastn hits were classified according to genera. Fig. 3 shows that the bronchial asthma samples were dominated by Streptococcus spp., Prevotella spp., Neisseria spp., and Veillonella species. The estimated diversity in the affected patients was 72.6%, whereas in the control group it was 46.6%. There were a higher number of bacterial genera represented in the normal control than in the bronchial asthma samples, including Granulicatella spp. (3.6%), Campylobacter spp. (2.6%), Capnocytophaga spp. (2.6%), Xylanibacter spp. (2.6%), Sphingomonas spp. (1.6%), Phocoenobacter spp. (1.0%), Schlegelella spp. (1.0%), Atopobium spp. (0.5%), Catonella spp. (0.5%), Dialister spp. (0.5%), Enterobacter spp. (0.5%), Gemella spp. (0.5%), Kaistella spp. (0.5%), Microvirgula spp. (0.5%), Novosphingobium spp. (0.5%), Oscillibacter spp. (0.5%), and Parvimonas spp. (0.5%). The presence of several genera in the bronchial asthma oropharynx samples separated these samples from the non-asthmatic control samples. For instance, Bergeriella spp., Megasphaera spp., Paraprevotella spp., Phycicola spp., Pilibacter spp. and Streptobacillus spp. were detected in the bronchial asthma oropharynx samples but were absent from the non-asthmatic control samples.
2-1-3. UniFrac and statistical analysis
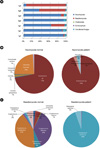
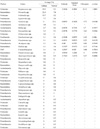
Jackknife clustering of environments (Fig. 4A), which is similar to principle component analysis (PCA), showed fairly robust clustering (75% bootstrap support for all nodes but one) in the bronchial asthma and non-asthmatic samples. Comparison of individual samples using Fast UniFrac PCA (Fig. 4B) showed a distinct clustering by dietary treatment when DNA samples from both groups were included in the analysis. Statistical analysis showed significantly differences in the frequencies of phyla between the bronchial asthma and non-asthmatic samples (Table 1). At the genus level, Neisseria spp. (p = 0.0168), Alcaligenes spp., Atopobium spp., Campylobacter spp., Catonella spp., Dialister spp., Enterobacter spp., Fusobacterium spp., Gemella spp., Granulicatella spp., Haemophilus spp., Hallella spp., Microvirgula spp., Novosphingobium spp., Oscillibacter spp., Parvimonas spp., Porphyromonas spp., Pseudomonas spp., Rothia spp., Schlegelella spp., Sphingomonas spp., Xylanibacter spp., and TM7 were abundant in the non-asthmatic individuals. In contrast, Streptococcus spp. and Anaerosporobacter spp. were predominant in the asthmatic patients at a significant level (p < 0.0001).
2-2. Diversity of fungal communities (Mycobiome) using next-generation sequencing
This study has been published in the PLOS ONE (36).
2-2-1. Background
Unlike classical seborrhoeic dermatitis, dandruff is a noninflammatory condition of the scalp that is characterized by scaling and is considered to be a form of mild seborrhoeic dermatitis. Dandruff is a common scalp disorder that affects almost half of the postpubertal population, regardless of ethnicity and gender, and has several putative causes including non-microbial and microbial factors (37, 38). Potential non-microbial causes of dandruff are excessive exposure to sunlight, irritation of the scalp due to over-shampooing, frequent combing, use of certain cosmetic products, and exposure to dust and dirt, although experimental evidence is lacking (37, 39, 40). The microbial etiopathology that is most widely accepted is the presence of a lipophilic yeast belonging to the genus Malassezia (37, 41). The scalp has a biotic community of which the known components are Staphylococcus spp., Propionibacterium spp., and Malassezia spp. (42). Dandruff-afflicted scalps have been reported to have 1.5-fold to 2-fold higher levels of Malassezia than non dandruff-afflicted scalps (43). Malassezia species are known to be the causative pathogens of pityriasis versicolor, seborrhoeic dermatitis (SD), and atopic dermatitis (AD) (44, 45). The genus Malassezia comprises at least seven species, each of which has specific ecological, biochemical, and genetic characteristics. Some Malassezia species (M. furfur, M. gobosa, and M. restricta) are associated with various human infections, but the pathological role of individual species is not yet fully understood (45~48). To investigate the fungal biota associated with dandruff, 26S rRNA gene sequences from fungal species collected from basal (healthy) scalp samples and dandruff-afflicted scalp samples were analyzed using the GS-FLX Titanium sequencer (Roche) for massively parallel ribosomal RNA gene amplicon pyrosequencing.
2-2-2. Comparison of fungal microbiota in the human scalp between dandruff-afflicted and non-dandruff-afflicted individuals
A total of 74,811 sequence reads of the 26S rRNA gene were obtained using the Genome Sequencer FLX Titanium platform (Table 2). Statistical analysis revealed that three phyla and 11 genera identified by sequencing and BLAST analysis had significantly different frequencies in healthy scalps and dandruff-afflicted scalps (Table 3). At the phylum level, Basidiomycota were isolated from dandruff-afflicted scalps twice as often as from healthy scalps. Sequences derived from fungi in the genera Acremonium, Coniochaeta, Cryptococcus, Didymella, Rhodotorula, and an uncultured Ascomycete were found in greater frequency in libraries generated from healthy scalp samples. In contrast, Eupenicillium, Filobasidium, Malassezia, and Penicillium were found significantly more often in samples from dandruff-afflicted scalps (p < 0.0001) along with an uncultured soil fungus (p = 0.0014). The most dominant fungal phylum in samples from the dandruff-afflicted scalps was Ascomycota, which accounted for 94.31%, 91.23%, 65.00%, 95.01%, 88.32%, 37.21%, and 6.28% of the fungal population of the seven swab samples, respectively, while Basidiomycota accounted for 2.32%, 1.55%, 32.26%, 0.02%, 7.06%, 61.66%, and 93.49% of the fungal population of each of the seven swab samples, respectively (Fig. 5A). A total of 53 species in the phylum Ascomycota were detected in the samples overall. Thirty-seven species were identified on healthy scalps and 24 were identified on dandruff-afflicted scalps. Eighteen species of Basidiomycota were detected on healthy scalps and 11 species were identified on dandruff-afflicted scalps. Acremonium spp. (phylum Ascomycota) was the most dominant fungus on both healthy and dandruff-afflicted scalps. On healthy scalps, Acremonium spp. and Didymella bryoniae comprised over 95% (72% and 23%, respectively) of the Ascomycota, which corresponds to >80% of the fungal community of healthy scalps compared to just over 60% in dandruff-afflicted scalps. In dandruff-afflicted scalps, >90% of the Ascomycota were Acremonium spp., and several Penicillium spp., including P. chrysogenum (2%) and P. meleagrinum (3%) were also present (Fig. 5B). In the phylum Basidiomycota, populations of Cryptococcus spp. observed on healthy scalps were replaced with Filobasidium (94%) on dandruff-afflicted scalps (Fig. 5C).
2-2-3. Discovery of an unusual fungus, Filobasidium floriforme
Filobasidium floriforme was present at a high frequency on the two scalps graded as having the most severe dandruff condition (5g5; 62.5% and 7g5, 92.9%; see Fig. 6). Even though their total frequency was lower, Penicillium species (P. chrysogenum and P. meleagrinum) showed the same tendency. Among the samples from healthy scalps, only 3g1 had a high frequency (32%) of species in the genus Cryptococcus, including C. albidosiilis (1.72%), C. diffluens (12.81%), and C. liquefaciens (14.7%). Several Malassezia spp., which are involved in pityriasis versicolor, SD, and AD as discussed previously (49), were detected in 2g1 and 4g4 scalps, including M. globosa, M. restricta, M. samuelsii, and one non-culturable species. Malassezia spp. were present in healthy scalp samples at a frequency of 0.07% and in dandruff-afflicted samples at a frequency of 2%. Filobasidium floriforme belongs to the phylum Filobasidiales. The Filobasidiales are an order in the fungal class Tremellomycetes. The one family in this order, the Filobasidiaceae, contains five species, namely F. capsuligenum ATCC 14437T, F. uniguttulatum ATCC 32047T, F. elegans, F. floriforme and F. globisporum (50). Filobasidium spp. have not been studied intensively in the context of human disease. Only one species, F. uniguttulatum, has been reported to cause cryptococcal meningitis (51). F. uniguttulatum is a teleomorphic fungus that was first isolated from an infected human nail in 1934 (52) and named Eutorulopsis uniguttulata. In 1952, it was renamed Cryptococcus neoformans var. uniguttulatus on the basis of morphological and physiological similarities with C. neoformans, but with reduced capsule formations. Phylogenetically, F. uniguttulatum is more closely related to C. albidus than to C. neoformans (53, 54). The results of the dandruff study suggested that the major fungus associated with dandruff is F. uniguttulatum rather than Malassezia spp. Therefore, control of Filobasidium spp. could potentially be an alternative strategy for treating dandruff.
SUMMARY
Metagenomic analysis is a valuable technique for studying microbial communities. In this review, I highlighted the potential applications of two types of metagenomic approaches to identify and detect causative organisms of unexplained diseases. These approaches involved the genomic analysis of microorganisms by Sanger sequencing and 454 pyrosequencing. These studies revealed the distribution and profile of bacteria and fungi associated with asthma and dandruff, respectively, thereby contributing to defining the microbial community associated with these disease states. The studies discussed above highlight how a metagenomic approach can be used to discover unknown microorganisms that may cause idiopathic infectious diseases.





 XML Download
XML Download