

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Flavivirus infection has resurged in recent decades and has caused hundreds of thousands of deaths annually worldwide (1~11). The genus Flavivirus of the family Flaviviridae is composed of more than 70 viruses that are transmitted by mosquitoes, ticks, or zoonotic agents with unidentified vectors (8). Approximately 40 viruses in the genus are associated with human diseases. Among these flaviviruses, dengue virus (DENV), West Nile virus (WNV), Japanese encephalitis virus (JEV), yellow fever virus (YFV), St. Louis encephalitis virus (SLEV), and tick-borne encephalitis virus (TBEV) are significant human pathogens globally, which induce severe encephalitic or hemorrhagic diseases that cause extensive morbidity and mortality (6, 8, 12~14).
Flaviviruses are single-stranded, positive-sense, enveloped RNA viruses with an approximately 11-kilobase genome. The genome is translated as a single polyprotein, which is cleaved by viral and cellular proteases to generate three structural proteins [capsid (C), pre-membrane (prM), and envelope (E)] and seven nonstructural (NS) proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) (15). Among them, the E protein functions in binding of the virus to a cell surface receptor, membrane fusion, and viral assembly with the assistance of prM (16). Flavivirus NS1 is a glycoprotein that is absent in the virion and secreted at high levels (up to 50 µg/ml) in the serum (17~24). Secreted NS1 associates with the cell surface membrane through interactions with sulfated glycosaminoglycans (25). Additionally, secreted and/or cell-associated NS1 is implicated in the pathogenesis and immune regulation of flavivirus infection in that it binds complement-regulatory factors (26~30). Two other NS proteins, NS3 and NS5, have been characterized as the viral protease/helicase and RNA-dependent RNA polymerase, respectively, which form a viral replication complex with other NS proteins (31, 32).
Although advances in anti-flaviviral drug discovery have progressed significantly, no therapeutic agent for flavivirus infection is currently approved for human use. Recent studies suggest that monoclonal antibody (mAb)-based therapy could be a promising alternative strategy (33~36). Several groups have generated protective mAbs against the E and NS1 proteins of flaviviruses (33, 34, 37~44). The structural E protein is used as the major antigenic target to raise neutralizing antibodies (33, 35, 36, 45). However, it is difficult to generate neutralizing antibody for variant viruses due to the high mutation rate of the flavivirus RNA-dependent RNA polymerase (46). Sub-neutralizing concentrations of anti-E antibody also have theoretical potential to cause antibody-dependent enhancement (ADE) of flavivirus infection (47~50). Another major antigen, the flavivirus NS1 glycoprotein, induces the production of non-neutralizing protective antibodies (34, 51~53). Although the protective mechanisms and binding regions of a few anti-NS1 antibodies have been demonstrated (24, 34, 54~56), most still remain to be characterized. On the other hand, several antiviral agents have been tested to overcome the drawback for anti-flavivirus antibodies (57~63). Some of these agents inhibit flavivirus infection and reduce the significant mortality and morbidity associated with the infections (62, 63).
This review provides an overview of the major antiflavivirus antibodies and other anti-flaviviral agents with particular attention to: [1] protective and/or therapeutic antibodies against flavivirus infections, [2] the inhibitory mechanisms of protective and/or therapeutic antibodies, and [3] other candidates for anti-flavivirus therapeutics.
I. Protective antibodies against flavivirus infection
Flavivirus infection induces the humoral immune response in the host and elicits the production of anti-flavivirus antibodies that could limit viral spread and burden (64~69). As expected, many studies demonstrate that passive administration of polyclonal or monoclonal antibodies against flavivirus proteins protects mice from lethal flavivirus infection (33, 34, 37~44), and B-cell-deficient mice are more vulnerable to infection (66). These findings indicate that antibodies are one of the major components involved in protection against flavivirus infection.
1. Neutralizing antibodies against the E protein
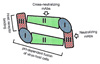
A flavivirus virion consists of three structural proteins (C, prM/M, and E), the viral genome, and a lipid envelope derived from the endoplasmic reticulum (ER) (70, 71). The viral particle induces the production of neutralizing antibodies (72~74). Although some neutralizing antibodies recognize the prM/M protein, the majority of neutralizing antibodies are raised against the E protein (75~77). The E protein has three structural domains and plays an important role in viral attachment, entry, assembly, and cell tropism (Fig. 1) (16). Domain I (DI) and Domain II (DII) of the E protein are involved in pH-dependent fusion of the virus and host cell membranes (78). Domain III (DIII) has an immunoglobulin-like fold and is located on the opposite end of DI, which is suggested to contain cellular receptor binding sites (43, 79, 80). Although neutralizing mAbs that cross-react with flaviviruses primarily recognize DII, most neutralizing antibodies bind to epitopes in the DIII region (Fig. 1) (43, 44, 81~83). In addition, X-ray crystallography and neutralization escape mutant analysis indicate that type-specific neutralizing antibodies against individual flaviviruses mainly map to amino acid residues in DIII of the E protein (Fig. 1) (84~87).
Interestingly, the anti-E neutralizing mAbs at sub-neutralizing concentrations have the potential to result in ADE of flavivirus infections, thereby complicating antibody therapy (47~50). Although ADE by anti-E neutralizing mAbs has not been well characterized in flavivirus infection in vivo, in vitro studies suggest that ADE is primarily associated with Fc-γ- or complement receptors (48, 88~90). To avoid the potential ADE, recent studies engineered anti-flavivirus antibodies with mutations in the Fc region, which prevented ADE in vitro and in vivo (88). Therefore, these findings suggest that ADE of flavivirus infection is a serious consideration in the design of novel strategies to develop a safe and effective therapeutic agent based on anti-E antibodies.
2. Non-neutralizing antibodies against the NS1 protein
During the course of natural infection, flaviviruses secrete nonstructural protein NS1, which is present at high concentrations (e.g., 1~50 µg/ml) in patient serum and associates with cell surface membranes (20~23, 25). Although most neutralizing antibodies are raised against virion-associated proteins, E and prM/M, NS1, which is absent from the virion, induces the production of non-neutralizing protective antibodies. Many studies have demonstrated the immunogenicity and protective efficiency of recombinant NS1 generated through DNA vaccines, recombinant viruses, and bacterial expression (38, 39, 51, 52, 91). The passive administration of anti-NS1 antibody protects mice against lethal flavivirus challenge, depending on the dosage and time of administration (34). These results suggest that anti-NS1 antibodies could serve as therapeutic antibodies that do not induce ADE. However, a recent study revealed that anti-DENV-2 NS1 mAbs (e.g., 1G5.3) cross-reacts with the DENV-2 E protein, inducing weak neutralizing activity and ADE in mice. These findings imply a potential risk of anti-NS1 antibody-based therapeutics (92).
Because mapping of protective mAbs could provide useful information for the design of effective therapeutics, several studies have been performed to determine the binding regions for anti-NS1 mAbs by using overlapping peptides, bacterially expressed fragments of NS1, yeast surface display expression, and mAb competition binding assays (24, 34, 54~56). Despite the intense interest, few protective mAbs against NS1 have been mapped to specific amino acids (53, 54, 91) and the three-dimensional structure of the NS1 protein has not yet been identified. Recently, important determinants for a cross-protective mAb against JEV and WNV, 16NS1, were identified by overlapping peptide mapping analysis combined with a yeast surface display system and site-specific mutagenesis (53). However, structural studies based on X-ray crystallography are required to further characterize the functions of flavivirus NS1 and protective anti-NS1 antibodies.
II. Protective mechanism of anti-flavivirus antibodies
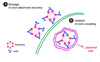
Protective antibodies prevent viral infection and reduce the viral burden in host cells through direct and indirect effects (33~35, 45). Neutralization is one of the direct functions of antibodies that does not require any other immune system components and is independent of the Fc portion of the antibody. Neutralizing antibodies block many steps in the viral entry pathway including virion attachment to the host cell, entry into the host cell, and uncoating in the endosome to release viral RNA into cytoplasm (Fig. 2) (93~96). For example, E53 and E60 anti-WNV E mAbs block viral attachment and entry at neutralizing concentrations (Fig. 2A) (96). The E16 anti-WNV E mAb inhibits fusion of WNV with the endosomal membrane and blocks uncoating, which leads the virus particle to the lysosome for destruction (Fig. 2B) (96). Although some neutralizing antibodies are well characterized, our understanding of neutralization is still limited. For example, an anti-DNEV2 mAb, 3H5-1, inhibits viral attachment to Vero cells, but the 3H5-1 mAb blocks DENV-2 fusion to the plasma membrane of LLC-MK2 cells (97, 98). In addition, while attachment factors (e.g., DC-SIGN, DC-SIGNR, heparin sulfate) that facilitate flavivirus binding in viral entry were identified (99~101), a neutralizing antibody that inhibits the E-attachment factor complex has not yet been characterized.
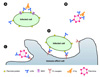
Non-neutralizing antibodies exert a protective effect through indirect functions that require the Fc portion of the antibody and components of the innate or adaptive immune system (102). Despite the absence of detectable neutralizing activity of the anti-NS1 antibody, many studies have reported its protective activity (34, 37, 39, 40, 42, 53, 91). Although the detailed mechanisms of this protection are incompletely understood, antiviral functions through an Fc-dependent pathway and complement-mediated cytolysis (CMC) of infected cells have been proposed as the basis for mAbs protection against NS1 (Fig. 3) (34, 42, 103). For example, anti-NS1 antibody bound to YFV-infected cells induces CMC of virus-infected cells (Fig. 3A) (103). Recent passive antibody transfer studies showed that anti-WNV NS1 mAbs (10NS1, 16NS1, and 17NS1) trigger protective activity through a C1q-independence and Fc-γ receptor I-and/or IV-mediated phagocytosis (Figs. 3C and D) (34, 37). However, specific mechanisms of many non-neutralizing antiviral antibodies against flavivirus NS1 remain to be identified. For example, an anti-WNV NS1 mAb, 14NS1, confers a strong protective effect in mice infected with lethal WNV that are deficient in C1q and Fc-γ receptor I and III (34), but the detailed mechanism underlying this effect remains uncharacterized. Although more studies on the detailed mechanisms of these anti-NS1 mAbs are needed, the facts that the cell surface-associated NS1 of WNV modulates complement activation by binding complement regulatory protein and the secreted NS1 of DENV increases viral propagation indicate mAbs to flavivirus NS1 may directly block the immunomodulatory and virologic functions of NS1 (26, 104).
Beyond direct neutralization of anti-flavivirus E mAbs, recent observations suggest that the protective activity of some neutralizing mAbs is partially dependent on the Fc portion of the mAbs and is associated with Fc receptors and the complement cascade (Fig. 3B) (48, 82, 105). For example, the protective efficiency of an anti-WNV E mAb is reduced in mice with blocked Fc-γ receptors I, III, and IV (82). The neutralization potential of hu-E16 anti-WNV E is augmented by C1q, which is dependent on the isotype of antibodies that bind C1q avidly (human IgG1 and IgG3) (48). Taken together, these recent findings suggest that a better comprehension of the role on protective antibodies in vivo and in vitro is crucial for the development of optimal therapeutic antibodies.
III. Other potential anti-flaviviral agents
Although antibody-based therapy provides a promising strategy to inhibit flavivirus infection, one possible limitation of this therapeutic antibody is the emergence of escape mutants that could decrease the inhibitory activity (46). Several antiviral agents have been tested and characterized to determine their inhibitory activity against viral replication in host cells (57~63). However, it is more difficult to develop specific antiviral agents without toxicity to cells because viruses, unlike bacteria, are obligate intracellular parasites that are dependent on the host's biosynthetic machinery. In this section, several anti-flaviviral agents will be discussed.
1. Interferons
Interferons are produced through the innate immune response to viral infection (106). During the viral life cycle, double-stranded viral RNA can primarily induce type I interferons such as interferon-α and interferon-β in the infected cell. Type I interferons inhibit viral replication by activating the JAK-STAT pathway, which induces the expression of antiviral genes. The interferon-dependent innate immune response is crucial for inhibiting flavivirus infection (107~111). For example, interferon-α/β receptor-deficient mice are more susceptible to WNV infection with 100% mortality and high viral loads in nearly all tissues (111). Type I interferon-pretreatment of cells also significantly inhibits flaviviruses (107~109, 111). However, the antiviral activity of interferon is significantly reduced after viral replication because flavivirus nonstructural proteins interfere with interferon signaling pathway (110, 112, 113). Nonetheless, several lines of evidence indicate that interferon has strong potential for use as a therapeutic. For example, treatment with interferon-α yields substantial improvement in complications in SLEV and WNV encephalitis cases (114, 115).
2. Nucleic acid-based inhibitors
Nucleic acid-based antiviral agents involve the use of oligonucleotides to suppress viral gene expression, including antisense-, ribozyme- and RNA interference (RNAi)-based approaches (116, 117). These strategies selectively inhibit viral replication by targeting the expression of key viral proteins through degradation of sequence-specific single-stranded RNA (118). RNAi is an evolutionarily conserved cellular mechanism that is initiated by double-stranded RNA (dsRNA) or micro RNA, which specifically blocks gene expression (119, 120). In the past decade, RNAi has been widely used to inhibit flavivirus infection in cells (57, 59, 121, 122). For example, small interfering RNA (siRNA) inhibits JEV replication (121). In addition, pretreatment of siRNA prior to viral replication significantly reduces WNV infection (59, 122). Recent studies show that administration of siRNA improves survival against lethal flavivirus infection in mice (62, 63). Although these results suggest that antiviral RNAi therapy is very promising, the emergence of escape mutations in the targeted sequences may limit their efficiency. However, the development of a new delivery method could increase their therapeutic potential and clinical applications because commonly used methods such as electroporation, lipid-based transfection reagents, and nanoparticles are less effective and not cell specific.
3. Small-molecule inhibitors
Viral replication is a well-organized process that is essential for effectively producing progeny viruses. Therefore, the steps of viral replication could be attractive targets for antiviral agents that inhibit viral genome replication and enzymes whose activity is crucial for viral protein processing (123). Two nonstructural proteins with enzymatic functions, NS3 (protease and helicase) and NS5 (RNA-dependent RNA polymerase), are considered as major targets for antiviral inhibitors, and a small-molecule library has been screened to identify compounds that block viral enzymes critical for replication (124~127). Borowski et al. demonstrated that an imidazo[4,5-d]pyridazine nucleoside analogue, 1-(2'-Omethyl-β-D-ribofuranosyl)imidazo[4,5-d]pyridazine-4,7(5H, 6H)-dione, inhibits the helicase activity of WNV NS3 (IC50 = 30 µM) (124). In addition, this nucleotide analogue shows similar inhibition against WNV replication in cell culture (125). Johnston et al. screened a compound library to identify inhibitors of WNV NS2B-NS3 protease using a miniature NS2B-NS3 assay and discovered a common amino-1H-pyrazol-3-yl scaffold as an inhibitor of WNV protease (126). Migliaccio et al. reported that nucleoside analogs, 2'-C-methyl-substituted ribonucleosides, inhibit flaviviruses such as WNV, DENV, and YFV by termination of RNA synthesis (127).
Several groups have recently used a high-throughput screening (HTS) assay combined with a flavivirus replicon and/or virus-like particle (VLP) system to discover novel flavivirus inhibitors (128, 129). Qing et al. developed an HTS assay using DENV-1 VLP to screen anti-DENV-1 inhibitors (128). Noueiry et al. employed a cell-based WNV subgenomic replicon to test over 80,000 compounds to determine their capacity to inhibit WNV replication and identified lead compound classes with strong antiviral activity against WNV, including 5H-cyclopenta [b] pyridine, tert-sulfonamide, thienylpyrimidine, and secondary sulfonamides (129). Approaches based on these HTS assays could facilitate the development of anti-flavivirus drugs that target viral entry, translation, and replication.
IV. Conclusions
During the past decades, the resurgence and spread of the most important mosquito-borne flaviviruses (e.g., DENV, WNV, and JEV) to new environments have increased the importance of developing anti-flavivirus agents. Anti-flaviviral candidates with different targets and inhibitory mechanisms have been developed. mAbs against the E and NS1 protein have provided significant protection through neutralization and effector-mediated clearance of virions and infected cells. In addition, other anti-flaviviral agents including type I interferons, oligonucleotide-based platforms, and small compounds targeting flaviviral replication have been developed to inhibit flaviviral infection in vitro and/or in vivo. Although these flavivirus antivirals demonstrate significant prophylactic and/or therapeutic effects against flaviviral infection, combination treatments are required to minimize the risk of escape mutant emergence and to increase the antiviral efficiency through different targets. Furthermore, expanding flavivirus epidemics and the co-circulation of multiple flaviviruses in the same endemic area (7~9) has heightened the necessity for developing broad-spectrum therapeutics against flavivirus infection.
 XML Download
XML Download