

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
It is well established and generally accepted that numerous innate and adaptive immune effector cells and molecules participate in the recognition and destruction of cancer cells, a process that is known as cancer immunosurveillance. However, cancer cells avoids such immunosurveillance through the outgrowth of poorly immunogenic tumor -cell variant (immunoselection, a process that is also known as immunoediting) and through immunosubversion (1,2) The immune system interacts with tumors throughout their development, and the consequences of this interaction have substantial implications for cancer therapy. Although some host responses may inhibit tumor growth and progression, the immune system can also promote cancer by provoking chronic inflammation and, in turn, elaborating factors that drive tumor growth, survival, and angiogenesis. Failure of immunity can predispose a person to tumor development (3). T cell-mediated immunosuppression has been observed for decades without clarification as to which factors was responsible for this observation. Recently, a number of publications showed that progressive tumor growth evokes generation of immunosuppressor cells and suppressor T cells (4,5). The identification of regulatory T (Treg) cells represents a milestone in the field of immuology and provides an explanation for T-cell-mediated immunosuppression (6-11). Although Treg cells were originally identified for their ability to prevent organ-specific autoimmune disease in mice, emerging evidence suggests that Treg cells play a pivotal role in tumor immunology and contribute to tumor growth and progression, thereby having an important impact on the outcome of cancer patients (3,12-17). Likewise, tumor-induced immune suppression could be responsible for the failure of many promising cell-base vaccine attempts (18) and other immune-based therapies against cancer (2,3,15,19-21). Tumor-induced immune suppression is caused by numerous mechanisms, many of which involve the accumulation of immune-suppressive infiltrates in the tumor microenvironment. One of the most potent and well-studied suppressive phenotypes found in the tumor microenvironment is the Treg cells. Increasing evidence shows that Treg cell may also play an important role in immune evasion mechanisms employed by cancer (17,18,22-24). Tumors may differentiate, expand, recruit, and activate Treg (tumor Treg) cells via multiple mechanisms and potently abrogate antitumor immunity (25,26). Fig. 1 shows a schematic diagram of tumor-mediated generation of Treg cells in tumor microenvironment. Tumor Treg cells may promote local tumor growth at tumor site, and may be relevant to the progression of systemic disease in the peripheral blood or lymphoid organs (25,27,28). Thus, tumor Treg cells may be a major obstacle for immunoterapy of cancer (25,27,29,30). Elucidating the mechanisms by which these cells traffic and accumulate in the tumor microenvironment could provide attractive therapeutic targets with which to combat tumor-induced immune suppression (11,31,32). An emerging interest is the potential role of Treg cells in cancer development and progression because they have been shown to suppress antitumor immunity (11,33). Thus, the critical role of Treg cells in tumor immunotherapy has been gradually elucidated (28,29,34,35). Treg cells (defined as CD4+CD25+FoxP3+ T cells) have been shown to be inversely related to the outcome of several human malignancies (26,36-41). Furthermore, depletion of Treg cells can enhance rejection of endogenous immune mediated tumor and improve tumor antigen-specific immunity, which highlights the impact of Treg cells in antitumor immunity (28,42,43) Depletion of Treg cells has also been shown to improve the efficacy of tumor immunotherapy, including vaccination and CTLA4 blockade (28-30,44).
In this review, I will discuss the role of human Treg cell in cancer. With this in mind, I briefly outline the evidence describing the role of Treg cell in impairing antitumor immune responses. And, I will review the recent studies demonstrating that Treg cells are increased in patients with malignancies. I will also review the effects of depletion or reduction of Treg cells on tumor progression and alterations of Treg homeostasis by cancer treatment. In addition, I will discuss both the prognostic value of Treg cells in tumor progression or survival of tumor-bearing animals and humans and the rationale for clinical use of Treg cells. It will emphasize the difficulties, as well as the opportunities, encountered in transitioning from bench to bedsides. I will confine this discussion to CD4+CD25+ or CD4+CD25+FoxP3+ Treg cells that develop in both the thymus and periphery and represent the major Treg cell populations that act critical for immune homeostasis (45). FoxP3-iTreg cells such as Tγ1 and Th3 cell will not be discussed here.
Treg CELLS
There has been an explosion of literature focusing on the role of these cells in several settings including cancer immunity, autoimmunity, transplantation tolerance allergic responses and microbial immunity (reviewed in 9,12-14,46-48). Treg cells can be described as a T-cell population that functionally suppresses an immune response by influencing the activity of another cell type (49,50). Treg cells were initially described by Gershon et al (6,7) in the early 1970s and were called suppressor T cells. Thereafter, a series of papers concerning thymic suppressor cell which could inhibit cell-mediated immunity in many different in vivo models (51-55). Unfortunately, despite the importance of these studies there was extensive skepticism in the immunological field about existence of these cell, and suppressor T cells left the center stage of immunology for decades. However, a rejuvenation of the Treg field was launched through the work of Sakaguchi et al (10,11) who in 1995 reported that a small group of T cells with particular cell surface phenotype (CD4+ cells which co-express the IL-2 receptor-α chain, CD25) maintain self-tolerance and that breakdown of this tolerance could lead to autoimmune disease and CD4+CD25+ T cells were named Treg cells. Now the terms "suppressor T cells" and "regulatory T cells" are used interchangeably, but the term "regulatory T cells" is preferred by most researchers. Work in the field of Treg cell immunology was greatly enhanced in 2003 by the discovery and characterization of the Treg-specific gene, FoxP3 (45,56-59). So far the most specific marker for naturally occurring CD4+ Treg cells, at least in mice, is FoxP3, a member of the forkhead family of DNA-binding transcription factor (45,56,58,59). More recent studies have shown that FoxP3 is not only a key intracellular marker but is also a crucial development and functional factor for CD4+CD25+ Treg cells (45,59-61). Until now, there are at least three recognized subsets of CD4+ Treg cells involved in the negative regulation of immune response, which include CD4+CD25+FoxP3+, type 1 Treg cells (Tγ1) secreting IL-10 and Th3 cells secreting TGFβ (62-66). Among these, classical Treg cells are CD4+CD25+FoxP3+ T cells and depletion of CD4+CD25+ FoxP3 cells has attracted much attention in recent years (28,29,58,67,68). Treg cells, according to their sources, broadly belong to two classes, which are naturally occurring thymic-derived Treg cells and adaptive or inducible Treg cells. Naturally occurring Treg (nTreg) cells are generated in the thymus during development of T lymphocytes (62,63,69). These Treg cells, constituting 5-10% total lymphocytes, then enter peripheral circulation and are widely distributed in peripheral reservoir lymph nodes and spleens (63,64,70). On the contrary, adaptive or inducible Treg (iTreg) cells are developed in the periphery from naive T cells or nTreg cells under the influence of various inductive signals, most importantly TGFβ or IL-10 (54,62,63,69,71). Treg cells are primarily characterized by CD4+FoxP3+ or CD4+CD25+FoxP3 T cells and FoxP3 has been considered as a master regulatory transcription factor for Treg cells (45,56,58,59). It has recently been reported that FoxP3+ Treg cells could also be generated outside the thymus under a variety of conditions (54,59,72). It has become evident that FoxP3+ Treg cells are one of the main barrier to implementation of cancer immunotherapies (12,21). Tumor tissue promote the conversion of naive T cells into FoxP3 Treg cells and accumulation of Treg cells in tumor site, thereby impairing the development of effector responses (33,60,61). Thus, FoxP3+ Treg cells hava a key function in obsructing tumor immunosurveillance (33,57,73). Although nTreg cells develop in the thymus, several reports have demonstrated that peripheral CD4+CD25+ T cells can be converted into adaptive FoxP3+Treg cells (74,75). Among the other major surface markers, glucocorticoid-induced TNF-receptors-related (GITR) protein and cytotoxic lymphocyte-associated angigen-4 (CTLA-4) are the most important with respect to development and function of Treg cells (45,47,48,69). Intrestingly, it has been reported that the large number of different cell types that are purported to be directly targeted by FoxP3+ Treg cells are CD4+, CD8+ T cell, dendritic cells, B cells, marophages, osteoblasts, mast cells, NK cells, and NKT cells (45).
INCREASED PREVALENCE OF Treg CELLS IN CANCER BEARING MICE
Recently, in murine models, a vast numbers of murine studies have shown that prevalence of Treg cells is increased in tumor bearing mice and the depletion or inhibition of Treg cells can enhance antitumor immunity (76-78). In context of cancer, Treg-cell frequencies and function are important because increased number might favor tumor development or growth and influence the course of the disease. It has been also shown that CD8+ T cell immunity against a tumor or self-antigen is hindered by Treg cells and Treg cells inhibit adoptive immunotherapy in pme-l TCR transgenic mice (77). In a mouse pancreas cancer model, tumor actively promotes the accrual of Treg through several mechanisms involving activation of nTreg cells as well as conversion of non-Treg into Treg cells (78). Both antitumor effector T cells and Treg cells capable of abrogating the antitumor reactivity of the effector T cells are primed in the same tumor-draining lymph nodes during tumor progression and the Treg cells generated in tumor-draining lymph nodes possess the same functional properties as the Treg cells that arise naturally in the thymus but recognize tumor-associate antigen (79). One study reported that the numbers of FoxP3+CD4+nTreg cells are significantly increased in draining but not non-draining, lymph nodes and spleen of tumor-bearing mice (80). In this study, furthermore, a small number of nTreg cells could be also found at the tumor sites. In a transgenic mouse model of prostate dysplasia, functional studies confirmed a role for CD4+CD25+ Treg cells in suppressing T cell proliferation as well as regulating the growth of transplanted prostate tumor cells (81). Additionally, these studies showed that anti-CD25 antibody treatment reduces, but does not prevent, tumor growth in a transgenic mouse model of prostate dysplasia, These transgenic mouse developed reproducible prostate specific tumors that can progress to mouse prostatic intraepithelial neoplasia similar to human prostatic intraepithelial neoplasia (81). Interestingly, in the mouse tumor model, no difference was observed in the proportion of CD4+CD25+/CD4+ in peripheral blood lymphocytes between colon-carcinoma-bearing BALB/c mice and normal mice, while there was a significant increase in the proportion in spleen lymphocytes in tumor bearing mice as compared with normal mice (82). Moreover, the proportion increased in accordance with the increase in the tumor size, suggesting that the CD4+CD25+/CD4+ proportion in spleen lymphocytes might be a sensitive index to evaluate the Treg cell in tumor mouse models. In one study, the transforming growth factor-β (TGFβ) secreting murine pancreas adenocarcinoma (Pan02) cell line was injected into syngeneic C57BL/6 mice. and the prevalence of Treg cells in the tumor draining lymph nodes (TDLNs) and spleen was measured weekly. Compared with control mice, the prevalence of CD4+CD25+ Treg cells in TDLNs and in tumor spleen increased with tumor growth, providing evidence that Pan02 tumor promotes the prevalence of Treg cells (32).
A study showed, using murine colorectal tumor, that tumor immunity stimulated in the absence of regulatory cells is not restricted to tumors of colorectal origin, but is effective against tumors of different histological types such as B cell lymphoma and a renal cell carcinoma (83). The data suggest that the generation of cross-reactive tumor immunity is a specific manifestation of CD25 Treg cell depletion and the generation of CD4+ T cells capable of mediating tumor rejection is another important feature of tumor immunity induced in the absence of CD25+ cells. Interestingly, Ghiringhelli et al investigated the mechanisms of immune tolerance raised by tumors by comparing immunogenic and tolerogenic tumor cell clones isolated from a rat colon carcinoma (84). When injected into syngeneic hosts, the immunogenic REGb cells yield tumors that are rejected, while the tolerogenic PROb cells yield progressive tumors and inhibit the regression of REGb tumors (84). They found that PROb tumor volume is correlated with an expansion of CD4+CD25+ Treg cells in lymphoid tissues. While total T cells from PROb tumor-bearing rats yield no apparent antitumor immune response, depletion of CD25+ T cells restores this reactivity. A single administration of cyclophosphamide depletes CD4+CD25+ Treg cells in PROb tumor-bearing animals, delays the growth of PROb tumors, and cures rats bearing established PROb tumor when followed by an imunotherapy which has no curative effect when administered alone. These data strongly suggest that role of Treg cells in tumor-induced immune tolerance and the interest of Treg cell depletion to sensitize established tumors to immunotherapy. Interestingly, furthermore, a recent study demonstrated the role of Treg cells in tumors of the central nervous system (85). The authors implanted syngeneic GL26 tumor cells in the brain of C57BL/6 mice, and tumor-infiltrating lymphocytes (TILs) was analyzed and found that CD4+CD25+ Treg cells play an important role in suppressing the immune response to tumors of central nervous system, suggesting that these Treg cells may therefore represent a potentially novel target for immunotherapy of malignant glioma. Schabowsky et al showed that Treg cells are actively recruited and induced by tumors to block innate and adaptive immune priming, effector function and memory response which can inhibit the efficacy of therapeutic cancer vaccines (18). Combination of CTLA-4 blockade and depletion of CD25+ Treg cells results in maximal tumor rejection (86). The synergism in the effects indicates that CD25+ Treg cells and CTLA-4 signaling represent two alternative pathways for suppression of autoreactive T cell immunity, suggesting that simultaneous intervention with both regulatory mechanisms is therefore a promising concept for the induction of therapeutic antitumor immunity.
It has been reported that cyclooxygenase (COX)-2 and its product prostaglandin (PG)E2 underlie an immunosuppressive network that is important in the pathogenesis of non-small cell lung cancer (86). Tumor-derived COX-2/PGE2 induced expression of the Treg cell-specific transcription factor, FoxP3, and increased Treg cell activity. In vivo, COX-2 inhibition reduced Treg cell frequency and activity, attenuated FoxP3 expression in TILs and decreased tumor burden, providing evidence that inhibition of COX-2/PGE2 suppresses Treg cell activity and enhances antitumor responses (87).
Concomitant tumor immunity describes the immune response in a host bearing a progressive tumor that rejects an inoculum of the same tumor at distant site (34,88). Progression of poorly immunogenic B16 melanoma did not spontaneously elicit concomitant immunity (88). However, depletion of CD4+ T cells in tumor-bearing mice resulted in CD8+ T cell-mediated rejection of challenge tumors. Concomitant immunity was also elicited by treatment of cyclophosmamide. nTreg cell efficiently suppressed concomitant immunity mediated by previously activated CD8+ T cells (88). These results shows that Treg cells are the major regulator of concomitant tumor immunity against weakly immunogenic tumor. In a series of elegant studies, most recently, Lin et al found that CD8+ T cells, from mice that lost concomitant tumor immunity, possessed potent antitumor properties and strongly expressed effector molecules (34). Furthermore, interestingly, effector/memory Treg cells increased as the primary tumor progressed. They also reported that these effector/memory Treg cells inhibit concomitant tumor immunity in vivo. Their data suggest that effector/memory Treg cells are responsible for the loss of concomitant tumor immunity associated with tumor progression.
Valzasina et al reported that tumor injection in thymectomized and CD25-depleted mice shows that CD4+CD25+ T-cell expansion occurs even in the absence of thymus (74). These newly generated cells are bonna fide Treg cells in terms of FoxP3 expression. In addition, they analyzed Treg cell number and function in tumor-bearing mice as well as their recovery after peripheral and central depletion by means of anti-CD25 monoclonal antibody (mAb) and thymectomy, and described CD4+CD25-T-cell conversion into Treg cells as the main mechanism of Treg cell replenishment and expansion in tumor-bearing mice.
Recently, there is a large number of evidence illustrating the increase in the number of Treg cells in the tumor setting, but very little work has been done to address the functionality of these cells. Despite the numerous studies showing that Treg cell from tumor-bearing animals and cancer patients are functional, no studies have directly compared Treg cells from tumor-bearing and nontumor-bearing animals. A recent study directly compared Treg cells in the spleen and the tumor infiltrate at both the molecular and functional levels (89). This study provides evidence that tumor-infiltrating Treg cells are functionally impaired, due to a loss of ability to respond to TCR stimulation and that murine Treg cells in the tumor microenvironment display both enhanced proliferation and reduced functionality. This enhanced proliferation leads to an intratumoral accumulation of Treg cells with a unique phenotype: CD4+CD25+FoxP3+GITRhighCD27llowCD621L-. The data further demonstrate that the accumulation of Treg cells in the tumor is caused by multiple factors, including increased proliferation, decrease apoptosis, and altered expression of chemokines, chemokine receptor, and cell-surface markers. The trafficking of T cells is mediated in part by chemokines and the expression of their receptors. Higher expression of the chemikine receptor CCR4 was reported on Treg cells. Likewise, CCR4 associated chemokines and macrophage-derived chemokine (CCL22) have been correlated with higher frequencies of Treg cells in gastric cancer (90). In ovarian cancer, tumor-derived CCL22 was shown to recruit Treg cells in vivo and predicts a negative prognosis (91). Most recently, Mailloux et al examined Treg cell levels and changes in chemokine secretion in a murine model of Lewis lung carcinoma (LLC) and demonstrated increased number of CD4+CD25+FoxP+ Treg cells in the lungs of C57BL/6 mice bearing a metastatic LLC (31).
It has been recently reported that in a syngeneic murine glioma model a time-dependent accumulation of CD4+FoxP3 Treg cell in brain tumor (92). Further analysis revealed a time-dependent upregulation of CD25, CTLA-4, GITR on intratumoral CD4+FoxP3+ Treg cells during tumor growth. Moreover, treatment with anti-CD25 mAbs significantly reduced the number of these highly suppressive Treg cells within the growing tumor and provoked a CD4 and CD8 T cell dependent destruction of the glioma cells. Combining Treg cell depletion with administration of blocking CTLA-4 mAbs further boosted glioma-specific CD4+ and CD8+ effector T cells as well as antiglioma IgG2a antibody titers resulting in complete tumor eradication (92). These data illustrate that intratumoral accumulation and activation of Treg cells act as a dominant immune escape mechanisms for glioma and underline the importance of controlling tumor-infiltrating Treg cells in glioma immunotherapy. Most recently, Liu et al have beautifully investigated the suppressor function of both tumor and naive Treg cells in vitro and in vivo and reported that Treg cells from mice bearing a breast tumor were elevated (tumor Treg cells) and that tumor Treg cells potently abrogate tumor-specific CD8+ T cell response in tumor-draining lymph nodes, thereby suppressing antitumor immunity at the early stage of the immune response induced by adoptively transferred tumor-primed CD4+ T cells (17).
RESTORATION OF ANTITUMOR IMMUNITY IN MICE BY DEPLETION OR REDUCTION OF Treg CELLS
As describe before, accumulating data have shown that Treg cells play an important role in tumor immunology. Increasing evidence for their role in fostering immune privilege in both mouse and human tumors has fueled interest in attempts to interfere with their number or function for therapeutic benefit (14,28). There is a large body of data in the literature demonstrating restoration of antitumor immunity by depletion or reduction of Treg cells. An earlier study demonstrated that elimination of CD25+CD4 T cells can abrogate immunological unresponsiveness to syngeneic tumor in vivo and in vitro, leading to spontaneous development of tumor specific effector cells as well as tumor-nonspecific ones (93). Using the weakly immunogenic MCA205 sarcoma and the poorly immunogenic B16/BL/D5 melanoma, an early study demonstrated that although the antitumor immunity enhanced by the depletion of CD4+CD25+ Treg cells is insufficient to eradicate tumors, it augments the sensitization of immune T cells in the draining lymph nodes, thus, facilitating adoptive immunotherapy (94). Prasad et al showed that depletion of Treg cells before vaccination considerably increased the ability of vaccinated mice to survive tumor challenge, and allowed the development of long-lasting tumor protective immunity (95). This study suggests that it is possible to generate effective immunotherapeutics against weakly immunogenic tumors by combining treatment with dendritic cells (DC) loaded with apoptotic tumor cells and depletion of Treg cells. Van Meirvenne et al (96) described mRNA electroporation as an efficient gene delivery method to introduce tumor-antigen into murine immature DC. They showed in this report that a significant improvement in cytotoxic T lymphocytes (CTL) response is obtained both in the primary and in the memory effector phases when Treg cells are depleted in vivo prior to immunization, providing the convincing evidence for the merit of Treg cell depletion prior to immunization with mRNA-electroporated DC. Onizuka et al investigated the effect of in vivo administration of anti-CD25 mAB on the growth of eight tumors, including five leukemias, a myeloma, and two fibrosarcoma derived from four different inbred mouse strains that grew progressively in syngeneic mice (97). They found that a single injection of less than 0.125 mg of anti-CD25 mAb caused a reduction in the number of CD4+CD25+cells and regression of six of the eight tumors, suggesting effective tumor rejection responses resulted from depletion of Treg cells. A study also showed that elimination of CD4+ T cells by anti-CD4 antibody caused regression of a methylcholantrene-induced sarcoma growing in syngeneic A/J mice (98). A CD4+ T-cell clone, designated T595B1, was also established to elucidate the characteristics of CD4+ suppressor T cells (98). Interestingly, one report has shown using a murine model of melanoma that treatment of mice with anti-CD25 mAB facilitates long-term CD4+ T cell-mediated tumor immunity through depletion of CD25+ Treg cells (99). Furthermore, depletion of CD25+ Treg cells allows the host mice to induce both CD4+ and CD8+ antitumoral responses following challenge of colon cancer cells (100). Simultaneous depletion of CD25+ and CD8+ cells with mAb revealed that tumor-specific CD4+ T cells were able to reject colon cancer cells, a MHC class II-negative tumor (100). Depletion of Treg cell alone or in combination wih a whole tumor cell vaccine promotes a tumor-specific immune response (76). Thus, strategies incorporating Treg cell depletion might improve the efficacy of cancer vaccines. In addition, the suppression of antitumor immunity by Treg cells occurs predominantly at the tumor site, and that intratumor depletion of CD4+ cells, even at a late stage of tumor development, can be an effective treatment for well-established cancers (101). IL-2-induced antitumor immunity is enhanced by Treg-cell depletion and is due to expansion of the tumor-infiltrating cytotoxic CD8+ T cell population. More recently, using B16/BL6, a transplantable murine melanoma model, Quezada et al demonstrated dichotomy between the effects of Treg cell depletion on tumor rejection dependent on whether depletion occurs before (prophylactic) or after (therapeutic) tumor engraftment (102). In these studies, failure to promote rejection with therapeutic depletion (i.e., depletion after tumor establishment, which is more relevant to the clinical seting) was not related to lack of Treg cell depletion, to elimination of effector T cells, or to a failure to enhance systemic antitumor T cell responses, but correlates with failure of effector cells to infiltrate the tumor and increase the intratumor ratio of effector T cells/Treg cell, suggesting an alternative, clinically applicable strategy for the treatment of established tumor. In fact, carefully timed depletion of Treg cells has been shown to enhance tumor immunity.
In a series of elegant experiments, Wrzesinski et al reported that removal of Treg cells by nonmyeloablative total body irradiation of tumor-bearing mice improved the function of adoptively transferred cells for new antitumor immunotherapies (103). Surprisingly, they also demonstrated that superior antitumor efficacy is achieved by further increasing the intensity of lymphodepletion to a level that required hematopoietic stem cells. A recent study also showed that removal of CD25+ Treg cells enhanced antitimor immunity against local growth of B cell lymphoma and that induction or expansion of Treg cells could be one mechanism by which the growing tumor evades immune surveillance in mice (104). Treatment of mice with low-dose of cyclophosphamide or anti-25 antibody to deplete Treg cells unmasked latent T-cell antitumor activity and tumor-located ligation of CD3 and CD28 can activate both innate (NKT cells) and adaptive (CD4+ and CD8+ T cells) responses to create a tumor-destructive environment to control tumor growth, but modulation of Treg cells is necessary to unmask local adaptive antitumor responses (105). A recent study showed that inducible and activated CD4+CD25+ Treg cells in the regional draining lymph nodes in mice suppress host local immunity during the growth of benzo[a]pyrene-induced forestomach carcinoma (106) and deletion of Treg cells can promote host local immunity to suppress tumor growth (106).
In a variety of disease models, the chemokine receptors thus far identified as allowing Treg cells to migrate to sites of disease more efficiently than effector cells include CCR4, CCR5, CCR6, CCR7 and CCR8 (91,107). In one most recent study, Tan et al hypothesized that CCR5-dependent chemotaxis is important for Treg cell migration into pancreatic adenocarinoma and that disruption of this signaling would result in decrease migration of Treg cell into the tumor, thereby slowing tumor growth (107). In addition, they showed that in human and murine pancreatic adenocarcinoma, Treg cells found within the tumor microenvironment express CCR5 and demonstrated that CCR5 ligand, such as CCL5, are produced by the tumor cells themselves. Subsequently, they showed in the murine model that reduced CCL5 expression by the tumor results in decreased Treg cell migration to the tumor and slowed tumor growth. This study suggests that Treg cell migration into tumor microenvironment is mediated by the CCL5-CCR5 axis, and that blockade of this pathway could represent a novel immunomodulatory strategy for the treatment of cancer.
It has been recently reported that reduction of Treg cells can be exploited to provoke effective tumor immunity. Two cardinal features of FoxP3+ Treg cells are that they constitutively express CTLA-4 which only happens after activation in other T cell subsets, and that FoxP3 controls the expression of CTLA-4 in Treg cells. CTLA-4 is a potent negarive regulator of T cell immune resposnes, as illustrated by CTLA-4 knockout mice (45,56,57). A recent study showed that a specific deficiency of CTLA-4 in Treg cells results in spontaneous development of systemic lymphoproliferation, fatal T cell-mediated autoimmune disease, and hyperproduction of immunoglobulin E in mice, and it also produces potent tumor immunity (108). Interestingly, Treg-specific CTLA-4 deficiency impairs in vivo and in vitro suppressive function of Treg cells, strongly suggesting that CTLA-4 is a key molecular target for controlling Treg cell-suppressive function in both physiological and pathological immune responses including tumor immunity, autoimmuniy and allergy (108).
INCREASED PREVALENCE OF Treg CELLS IN PATIENTS WITH MALIGNANCIES
T cells with regulatory function were reported in patients with cancer even in the early 1990s (12-14). However, these reports were not followed until the identification of CD4+CD25+ Treg cells in 1995 (37). The first paper indicating the presence of CD4+CD25+ Treg cells in human tumor samples emerged in 2001 when Woo et al reported when observed increased percentages of CD4+CD25+ cells from patients with lung or ovarian cancer (109,110). Since then, there has been an explosion of literature demonstrating the presence of CD4+CD25+ Treg cells in patients with a variety of cancers (see Table I). Treg cells play an important role in the prevention of antitumor immune responses (15,21,64,93,111-113). The number of Treg cells in peripheral blood increases in many human cancers. The presence of the Treg cells is also likely to hinder the development of antitumor immune responses following the delivery of an immunotherapeutic agent. For this reason, methods of abrogating the activity of Treg cells may be critical for the successful immunotherapeutic treatment of cancer (89). As such, modulation of Treg cell functions in cancer has been studied using various approaches, with encouraging preclinical and clinical findings. A clear understanding of how immune function is modulated by tumor cell is required to devise strategies to enhance the efficacy of such therapeutic intervention.
Treg cells in solid tumors
It has been shown that patients with gastrointestinal malignancies had a higher proportion of CD4+CD25+ T cells in peripheral blood (114,115). Among patients with gastric carcinoma, those with higher percentages of CD4+CD25+ T cells had a poorer prognosis than did those with lower percentages. Interestingly, CD4+CD25+ T cells also were present in greater proportions in ascites from patients who had advanced-stage disease with peritoneal dissemination (114,115). A recent study showed that the percentages of CD4+CD25high T cells in peripheral blood mononuclear cells (PBMCs) from patients with gastric and esophageal cancer were significantly higher than those for healthy donors (116). Interestingly, after patients received curative resections of gastric cancer, the increased proportions of Treg cells were significantly reduced, and the levels were almost equal to those in normal healthy donors. A study also showed that the prevalence of peripheral blood CD4+CD25+ Treg cells in both gastric and esophageal cancer patients was significantly higher than that in healthy donors. The population of Treg cells in the TILs of gastric cancer patients with advanced disease was significantly higher than that in TILs of patients with early-stage disease (117). Most recently, it has been shown that prevalence of Treg cells in the peripheral blood of gastrointestinal cancer patients is significantly higher than that in healthy volunteers, even in the early stages of the disease (23). Moreover, since Treg cell levels are significantly reduced after curative resection, it is possible that tumor cells may have induced and expanded the Treg cell pool (23). Currently, Shen et al have characterized CD4+CD25CD127low/- as the surface marker of Treg cells in gastric cancer (16). They found that the frequency of CD4+CD25+CD127low/- Treg cells in the peripheral blood of gastric cancer patients was significantly higher than that in healthy controls. Increased Treg cells were also present in the tumor environment, such as those found in the ascites fluid, tumor tissue or adjacent lymph nodes. They proposed that CD4+CD25CD127low/- can be used as a selective biomarker to enrich human Treg cells and also to perform functional in vitro assay in gastric cancer. Currently, Xu et al also have investigated the prevalence of Treg cells in gastrointestinal cancer patients treated with chemotherapy and demonstrated that not only was the prevalence of Treg cells in the peripheral blood of gastrointestinal cancer patients significantly higher than that in healthy donors, but it also increased in parallel with tumor progression (118). In addition, among patients with advanced disease, 3 weeks after chemotherapy, those who higher percentage of Treg cells had a poorer prognosis. Recently, Mizukami et al investigated the frequency of CD4+CD25+FoxP3+ Treg cells in TILs, tumor-draining regional lymph nodes, and PBMCs of patients of gastric cancer, and evaluated the relationship between the presence of CCL17- or CCL22-producing cells and found that CC17 and CCl22 within the tumor are related to the increased population of FoxP3+ Treg cells, with such an observation occurring in early gastric cancer (90). Most recently, some studies demonstrated that CCL22 chemokines derived from a tumor induce the migration of Treg cells through CCR4, which is chemokine receptor for CCL22, and impair antitumor immunity in primary breast cancer and lung cancer (119,120). In patients with gastrointestinal cancer, as described previously, it has been shown that patients with gastrointestinal malignancies had a higher proportion of CD4+CD25+ T cells in peripheral blood (114,117). Wieczorek et al also found that Treg cell numbers are significantly increased in the peripheral blood of patients with IL-2-treated melanoma and in formalin-fixed tissue from patients with lung and colon carcinoma (121). In addition, they also demonstrated that Treg cell numbers are predictively elevated in the peripheral blood of patients with various solid tumors. Patients with squamous cell carcinoma of the head and neck have increased number of CD4+CD25+FoxP3+ Treg cells in their peripheral circulation compared with normal controls and have a depressed antitumor immnity (122,123). In addition, surprisingly, higher Treg frequency and levels of suppression were observed in patients with no evident disease than in untreated patients with active disease (122). Of interest, a recent study showed that patients with hepatocellular carcinoma also have increased numbers of CD4+CD25+ Treg cells in their peripheral blood, suggesting the increase in frequency of Treg cells might play a role in modulation of the immune response against hepatocellular carcinoma and could be important in design of immunotherapeutic approaches (37). Moreover, Treg cells are associated with hepatocellular carcinoma invasiveness, and intratumoral balance of regulatory and cytotoxic T cells is a promising independent predictor for recurrence and survival in hepatocellular carcinoma (124). It has been also demonstrated that primary hepatic cancers develop in liver that is immunosuppressed by a marked infiltration of CD4+CD25+FoxP3+ Treg cells. A high prevalence of Treg cell infiltrating hepatocellular carcinoma cells is thought to be an unfavorable prognostic indicator (125).
One line of study showed that measurement of heavy (H)- and light chain(L)-ferritin by ELISA revealed that H-but not L-ferritin was elevated in the circulation of melanoma patients and that the ratio of H-ferritin:L-ferritin correlated with the levels of Treg cells. In addition, this study revealed a marked increase in the number of CD4+CD25+CD69- T cells in such patients (126). Several studies showed that CD4+CD25high FoxP3+ Treg cells are overrepresented in human metastatic lymph nodes with a 2-fold increased frequency compared with both tumor-free lymph nodes (127-130) and that advanced melanoma is associated with increased numbers of circulating Treg cells and DCs and suggested that melanoma induces immunosuppressive DCs and Treg cells in the systemic circulation of the patients (131). Vence et al also showed the presence of tumor antigen-specific CD4+ Treg cells in the blood of patients with metastatic melanoma (132). These Treg cells recognize a broad range of tumor antigens, including gp100 and TRP1 (melanoma tissue differentiation antigens), NY-ESo-1 (cancer/testis antigen) and survivin (inhibitor of apoptosis protein family antigen). They suppress auatologous CD4+CD25- T cell responses in a cell-contact dependent manner. Such tumor antigen-specific Treg cells were not detected in healthy individuals. Thus, these tumor antigen-specific Treg cells might represent another target for immunotherapy of metastatic melanoma.
Interestingly, signal transducers and activation of transcription (STAT)5 and STAT3 oppose one another in regulation of the reciprocal development of CD4+CD25+FoxP3+ Treg cells and a reduction in STAT3 is associated with up-regulation of Treg cells, and STAT5 activation promotes Treg cell differentiation or function (133,134). The effects of IFNα on STAT signaling in relation to tumor Treg cells were not documented in humans beyond the observation that IFNα2b downregulates STAT3 (133,134). Wang et al investigated the effect of high-dose IFNα2b on regional lymph node metastases of human melanoma (134). They found that IFNα2b upregulates STAT5 and downregulates STAT3, in conjunction with upregulation of Treg cells in melanoma tissues, suggesting the effects of IFNα may be potentiated through interference with the response of Treg cells and/or STAT5. A recent study showed that administration of high dose IL-2 in patients with metastatic melanoma and renal cell cancer increased the frequency of circulating CD4+CD25+FoxP3+ Treg cells (135). These studies suggest that selective inhibition of IL-2-mediated enhancement of Treg cells may improve the therapeutic effectiveness of IL-2 administration. Nicholaou et al most recently showed in patients with melanoma that although strong antibody responses were mounted, the generation of delayed-type hypersensitivity response was significantly impaired and patients with advanced melanoma had a significantly higher proportion of circulating CD4+CD25+FoxP3+ Treg cells compared with those with minimal residual disease (20). The prevalence of Treg cells in the peripheral blood of pancreas cancer patients is increased when compared with normal individuals. Similarly, Treg cells are present in TILs and regional lymph nodes infiltrated by tumor. In addition, a recent study showed that the prevalence of Treg cells was significantly increased in the ductal adenocarcinomas compared with that in the stroma of nonneoplastic inflammation and the increased prevalence of Treg cells was significantly correlated with certain clinicopathologic factors (136). They also showed that a better prognosis was observed in patients with a low prevalence of Treg cells and that in pancreatic ductal carcinoma, a high prevalence of Treg cells seems to be a marker of poor prognosis.
In patients with prostate cancer, the prevalence of CD4+CD25high Treg cells inside the prostate was significantly higher in the tumor compared with benign tissue from the sam prostate (137). In this study, furthermore, the frequency of CD4+CD25high T cells in peripheral blood was significantly higher in prostate cancer patients compared with normal donors. Moreover, CD4+CD25+ T cells from blood and supernatants from cultured prostate tumor tissue samples exhibited immunosuppressive function in vitro. These findings indicate that Treg cells are an important cellular component of early-stage prostate tumors, and thus new therapeutic strategies aimed at inhibition or depletion of Treg cells may improve prostate cancer immunotherapy. Interestingly, a more recent study showed that although levels of CD4+CD25highFoxP3+ Treg cells in the peripheral blood of patients with prostate cancer were not significantly higher than those in healthy donors, Treg cells in patients with prostate cancer had significantly greater suppressive functionality than Treg cells from heathy donors (24). Additionally, there was a direct correlation between the serum levels of PGE2 and Treg cell functionality in patients with localized prostate cancers. Most recently, it has been reported that more than four hundred prostate cancer patients have elevated numbers of circulating and tumor infiltrating Treg cells and Treg cells increase tumor growth in vivo and these Treg cells potently inhibit tumor-specific T cells (138). In patients with renal cancer, interestingly, administration of high dose IL-2 in patients with renal cell cancer increased the frequency of circulating CD4+CD25+Foxp3+ Treg cells (135). These studies suggest that selective inhibition of IL-2-mediated enhancement of Treg cells may improve the therapeutic effectiveness of IL-2 administration. More recently, a study showed that it is the FoxP3- subset of CD4+CD25+ T cells and not the FoxP3+ subset that correlated with worse pathologic feature of renal cell carcinoma and cancer-specific survival (40). Most recently, Jensen et al also reported that intratumoral FoxP3+ Treg cells significantly increased during IL-2-based immunotherapy, and high numbers of on-treatment FoxP3+ cells were correlated with poor prognosis in patients with metastatic renal cell carcinoma (139).
In patients with ovarian cancer, tumor-associated T cells from patients with late-stage ovarian cancer contain increased proportion of CD4+CD25 T cells and may be contributing to T-cell immune suppression (109). In addition, a study also showed that there is an increasing of the proportion of CD4+CD25+ Treg cells in PBMC, TIL and tumor associated lymphocytes of the patients with ovarian carcinoma (140). In a large scale study of 104 patients with ovarian carcinoma, CD4+CD25+FoxP3+ Treg cells were shown to accumulate in tumors, in which they suppressed tumor-specific T cell immunity (91). In this study, most notably, the numbers of Treg cells present within tumor biopsy specimens of different patients was highly inversely correlated with patient survival. Tumor cells and microenvironmental macrophage produce the chemokine CCL22, which mediates trafficking of Treg cells to the tumor. These studies provide evidence that this specific recruitment of Treg cells represents a mechanism by which tumor may foster immune privilege and blocking Treg cell migration or function may help to defeat human cancer (91). Most recently, one line of evidence showed that disease specific survival was not influenced by the presence of FoxP3+ Treg cells in ovarian tissue, but median disease specific survival of patients with a high CD8+/FoxP3+ ratio in ovarian-derived tumor tissue was twice as high as for patients with a low CD8+/FoxP3+ ratio (141). Possibly, the positive prognostic effect of the of FoxP3+ cells in their samples can be attributed to the staining of not only suppressive, but also activated T cells expressing FoxP3. In patients with cervical carcinoma, all TDLN contained Treg cells whose frequency and suppressive function was not influenced by the neoadjuvant therapy (142,143). These studies showed that neoadjuvant therapy produces an enhancing effect on the immune competency of TDLNs from patients with cervical carcinoma. In endometrial cancer patients, similarly, Treg cells were observed in TDLN (143).
Of great interest, in malignancies of intrinsic central nervous system, Heimberger et al have recently shown that Treg cells were not present in normal brain tissue and were very rarely found in low-grade gliomas and oligodendrogliomas (41). They observed significant differences in the prevalence of Treg cells between astrocytic and oligodendorglial tumors, between different pathologic types of tumors. In addition, they identified Treg cells most frequently in glioblastoma multiforme but very rarely in low-grade astrocytomas. Furthermore, they also observed that the presence of Treg cells within glioblastoma multiformes did not alter the median survival in patients from whom the tumors were obtained. These results strongly suggest that Treg infiltration differed significantly in the tumors according to lineage, pathology, and grade, and that Treg cells seemed to have the highest predilection for tumors of the astrocytic lineage and specifically in the high-grade gliomas, such as glioblastoma multifmorme. The authors noted that the presence of Treg cells in glioblastome multiformes seemed to be prognostically neutral. A study also showed that the prevalence of peripheral blood CD4+CD25+ Treg cells in esophageal cancer patients was significantly higher than that in healthy donors (117). In addition, Kono et al showed that the percentages of CD4+CD25high T cells in PBMCs from patients with esophageal cancer were significantly higher than those for healthy donors (116). One study provided evidence of an increased pool of CD4+CD25+ Treg cells in the peripheral blood of cancer patients, including lung cancer, breast cancer, colorectal cancer and gastiric cancer patients with potent immunosuppressive features (144). In patients with lung cancer Woo et al showed that lung tumors contain large numbers of CD4+CD25+ cells and they have constitutive high-level expression of CTLA-4 (110). Furthermore, the CD4+CD25+ T cells mediate potent inhibition of autologous T cell proliferation, suggesting they could contribute to the progression of lung cancer. Wieczorek et al found that Treg cell numbers are significantly increased in the peripheral blood of patients with IL-2-treated melanoma and in formalin-fixed tissue from patients with lung and colon carcinoma (121). In addition, they also demonstrated that Treg cell numbers are predictively elevated in the peripheral blood of patients with various solid tumors. Most recently, Qin et al showed that pleural fluid from lung cancer patients was chemotactic for Treg cells, and intrapleural administration of the chemokine CCL22 of patients produced a marked progressive influx of Treg cells into pleural space, suggesting that CCL22 directly induce Treg cell infiltration into the pleural space in patients with malignant effusion (120). Tumor-associated T cells from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer contain increased proportion of CD4+CD25 T cells and may be contributing to T-cell immune suppression (109).
In patients with breast cancer, the prevalence of Treg cells in the peripheral blood of breast and pancreas cancer patients is increased when compared with normal individuals. Similarly, Treg cells are present in TILs and TDLNs infiltrated by tumor. These cells secret IL-10 and TGFβ and prevent activated CD4+CD25- and CD8+ from proliferating (36). Quantification of FoxP3+ Treg cells in breast cancer is valuable for assessing disease prognosis and progression, and Treg cells are an important therapeutic target for breast cancer. FoxP3+ Treg cells represent a novel marker for identifying late-relapse patients (145). Most recently, immunohistochemical analysis of FoxP3 in primary breast tumors showed that a high number of tumor-infiltrating Treg cells within lymphoid infiltrates surrounding the tumor was predictive of relapse and death (119). These Treg cells could be selectively recruited through CCR4. A most recent study also showed that Treg cell are involved in tumor onset and progression in human primary breast cancer, possibly contributing to poor prognosis of patients with breast cancer (146). A recent study elegantly showed that breast cancer patients have a greater percentage of circulating Treg cells along with serum levels of TGFβ than normal subjects, and that HER2/neu peptide (E75) vaccination increased circulating activated CD4+ T cells post-vaccination (147). However, Treg cells were significantly reduced after vaccination. Furthermore, activated Treg cells increased. Importantly, post-vaccination decreases in Treg cells were temporally associated with increased E75 vaccine-specific CD8+ T cells and corresponding HER/neu+ tumor cytotoxicity. These studies suggest that successful cancer vaccination strategies may require the alteration of complex immune interaction. Most recently, a study demonstrated that FoxP3 expression in breast cancer was associated with worse overall survival probability and the risk increased with increasing FoxP3 immunostaining intensity. FoxP3 was also a strong prognostic factor for distant metastases-free survival. The probability of 10-year survival in node-negative subgroup was 100% for FoxP3-negative and 82% for FoxP3-positive patients. Authors proposed that FoxP3 expression as a new independent prognostic factor in breast carcinoma, which might help to improve the selection of patients for appropriate therapy (148).
Treg cells in hematologic malignancies
The Wilms' tumor gene exerts an oncogenic function in various types of leukemias (149). It is also overexpressed in several solid tumors, and therefore, it has been considered as an attractive targer for cancer immunotherapy (149). Recently, a study demonstrated that a human HLA-restricted CD4+FoxP3+ Treg population has been detected in acute myeloid leukemia patients, suggesting that Wilms' tumor may be a novel target for leukemia-specific CD4+ Treg cells (149). Beyer et al demonstrated a significantly increased frequency of CD4+CD25highFoxP3+ Treg cell with strong inhibitory function in the patients with monoclonal gammopathy of undetermined significance (MGUS) or multiple myeloma (150). Prabhala et al also observed a significant increase in CD4+CD25+ T cells in patients with MGUS and in patients with multiple myeloma compared with healthy donors (151). However, Treg cells are significantly decreased in patients with MGUS and multiple myeloma compared with healthy donors. Moreover, even when they are added in higher proportions, Treg cells in patients with MGUS and multiple myeloma are unable to suppress anti-CD3-mediated T-cell proliferation (151). This decrease number and function of Treg cells may account, at least in part, for the nonspecific increase in CD4+CD25+T cells, thereby contributing to dysfunctional T-cell responses. In patients with adult T cell leukemia/.lymphoma (ATLL), FoxP3 expression was observed in ATLL cells (151,153). In patients with chronic lymphocytic leukemia, increased frequencies of CD4+CD25high Treg cells have been recently described as an additional mechanism reducing immunity in many patients with cancer (154). More recently, Beyer et al assessed 73 patients with chronic lymphocytic leukemia and demonstrated significantly increased frequencies of CD4+CD25highFoxP3+ Treg cells, with highest frequencies in untreated or progressing patients presenting with extended disease (155). Most surprisingly, in the majority of patients with chronic lymphocytic leukemia treated with fludarabine-containing therapy regimens the inhibitory function of Treg cells was decreased or even abrogated (155). In addition, frequencies of Treg cells were significantly decreased after therapy with fludarabine. The authors have proposed that in light of similar findings for cyclophosphamide the combination of fludarabine and cyclophosphamide might be further exploited in strategies reducing immunosuppression prior to cancer immunotherapy (155). In patients with acute myeloid leukemia, the frequency of CD4+CD25high Treg cells in peripheral blood is significantly higher when compared with healthy individuals. Notably, Treg cells in patients with acute myeloid leukemia presented significantly higher apoptosis and proliferation than that of health individuals (156). Most recently, it has been reported that Treg cell accumulating in the peripheral circulation of acute myeloid leukemia patients mediate vigorous suppression via contact-dependent and contact-independent mechanisms (157). Patients with lower Treg cell frequency at diagnosis have a better response to induction chemotherapy. Unexpectedly, patients who achieved complete remission still had elevated frequency of Treg cells, which mediated high levels of suppressor activity, suggesting that Treg cell are resistant to conventional chemotherapy (157). Accumulating data have provided that the presence of a high number of Treg cells in the solid tumors has been correlated with a poor prognosis of patients (25,38,141). However, most of these studies have been performed on carcinomas, with the role of Treg cells in hematologic malignancies such as non-Hodgkin lymphoma being less established. Contrary to these observations, Carreras et al reported that follicular lymphoma patients with higher Treg cell numbers in their tumors had a better response to therapy and improved overall survival (158). In this series of follicular lymphoma patients, Follicular Lymphoma International Prognostic Index (FLIPI) effectively segregated the patients into prognostically relevant groups. Interestingly, the prognostic value of the number of Treg cells was independent of the FLIPI score, suggesting that this parameter reflects a different biologic aspect of the tumors. These studies suggest that the role of Treg cells in the pathogenesis of these B-cell lymphomas may be different than carcinomas. Most non-Hodgkin lymphomas are of B-cell origin, but the tumor tissue can be variably infiltrated with T cells. A recent study showed that a subset of CD4+CD25+FoxP3+with high level of CTLA-4 are identified in biopsy specimens of B-cell non-Hodgkin lymphoma and these cells suppressed the proliferation and cytokine (IFN-γ and IL-4) production of infiltrating CD4+CD25- T cells in response to phytohemagglutinin stimulation (159). Using a human model of B-cell non-Hodgkin's lymphoma, Yang et al showed that intratumoral Treg cells inhibit the proliferation and granule production of activated autologous infiltrating CD8+ T cells (160). Moreover, their results showed that degranulation and subsequent cytotoxic acivity of infiltrating CD8+ T cells exposed to lymphoma B cell is completely attenuated by the presence of intratumoral Treg cells. In patients with Hodgkin's lymphoma, Hodgkin lymphoma-infiltrating lymphocytes are highly enriched for Treg cells, which induce a profoundly immunosuppressive environment for the ineffective immune clearance of Hodgkin-Reed Sternberg cells (161). Alvaro et al examined the expression of granzyme B and TIA-1 (markers for cytotoxic T lymphocyte) and FoxP3 independently by immunochemistry in tissue microarray of classic Hodgkin's lymphoma patients and correlated with patient outcome (162). They found that low infiltration of FoxP3+ cells in conjunction with high infiltration of TIA-1+ cells in classic Hodgkin's lymphoma and this result may represent biological markers predicting an unfavorable outcome. In line with a previous study (161), the presence of FoxP3+ Treg cells was detected in the reactive background of their classic Hodgkin's lymphoma patients.
EFFECTS OF DEPLETION OR REDUCTION OF Treg CELLS ON HUMAN TUMOR PROGRESSION
A recent study showed that TILs isolated from patients with hepatocellular carcinoma are compromised and contain a subpopulation of suppressive CD4+CD25+FoxP3+ Treg cells and that functional deletion of tumor-infiltrating Treg cells could enhance tumor-specific immunotherapy (163). Human CD4+CD25+ Treg cells has been isolated from human peripheral blood, thymus, lymph nodes and cord blood. In general, the characteristics of this T cell subset are strikingly similar between mouse and man (164). Depletion or blockade of Treg cells can also enhance immune protection form tumor-associated antigens that are expressed as self antigens (165). A most recent study showed that the relative increase in CD4+CD25+ Treg cells may be related to immunosuppression and tumor progression in patients with gastrointestinal cancer (118). In addition, chemotherapy in patients with advanced disease showed that immunosuppression is enhanced early (about 1~2 weeks) after chemotherapy, and increased Treg cells 3 weeks after chemotherapy correlated with a poor prognosis.
An early study showed that the IL-2 diphtheria toxin conjugate DAB389IL-2 (denileukin diftitox, ONTAK) have shown clinical activity in a variety of diseases, including B-cell non-Hodgkin's lymphoma, cutaneous T-cell lymphoma, Hodgkin's disease, psoriasis, rheumatoid arthritis, and HIV infection (166). Moreover, the highest response rates were observed in cutaneous T-cell lymphoma (166). This drug, denileukin diftitox has been approved by the Food and Drug Administration in the United States for treatment of CD25+cutaneous T-cell leukemia and lymphoma (167,168). Dannull et al used denileukin diftitox to selectively eliminate CD25-expressing Treg cells from cancer patients (169). This drug significantly reduced the number of Treg cells present in the peripheral blood of metastatic cell carcinoma patients and abrogated Treg cell-mediated immunosuppressive activity in vivo. Moreover, they showed that this drug-mediated elimination of Treg cells followed by vaccination with RNA-transfected DCs significantly improved the stimulation of tumor-specific T cell responses in metastatic renal carcinoma patients when compared with vaccination alone. Their results may have implications in the design of immune-based strategies that may incorporate the Treg depletion strategy to achieve potent antitumor immunity with therapeutic impact. It has been approved for treatment of patients with persistent or recurrent cutaneous T-cell lymphoma whose malignant cells express the CD25 component of the IL-2 receptor (166,168). A more recent study indicate that denileukin diftitox has activity in the setting of not only T-cell but also B-cell malignancies (167). A recent study showed that denileukin diftitoxin was a well-tolerated treatments in early- and advanced-stage cutaneous T-cell lymphoma and was not associated with detrimental immunologic effects on lymphocyte populations (168). Recently, there was a report demonstrating that the human IL-2 recepotrs exist in three isoforms such as low, intermediate, and high affinity (170). A number of leukemias and lymphomas, including cutaneous T-cell lymphoma, chronic lymphocytic leukemia, and B-cell non-Hodgkin lymphoma, express a component of the receptors (166). Ex vivo studies have shown that denileukin diftitox interacts with the high-and intermediate-affinity IL-2 receptors on the cell surface and undergoes internalization (170). Subsequent cleavages in the endosome releases the diphtheria toxin into the cytosol, which then inhibits cellular protein synthesis, resulting in rapid cell death (29,166,169). The clinical profile and potential benefits of denileukin diftitox in the treatment of cutaneous T-cell lymphoma were shown (170). More recently, a study provided the direct evidence that circulating CD4+CD25highFoxP3+ Treg cells are depleted after multiple doses of immunotoxin, denileukin diftitox and indicated the potential for combining Treg cell depletion with anticancer vaccines to enhance tumor antigen-specific immune response (29). In a series of elegant experiments, Litzinger et al recently reported that Treg cells in spleen, peripheral blood, and bone marrow of normal C57BL/6 mice were variously reduced after a single intraperitoneal injection of denileukin diftitox (171); the reduction was evident within 24 hours and lasted approximately 10 days. Injection of denileukin diftitox 1 day before vaccination enhanced antigen-specific T cell responses above levels induced by vaccination alone. These studies demonstrate in murine model (1) the differential effects of denileukin diftitox on Treg cells in different cellular compartments, (2) the advantage of combining denileukin diftitox with a vaccine to enhance antigen-specific T-cell immune responses, (3) the lack of inhibition by denileukin diftitox of host immune responses directed against a live viral vector, and (4) the importance of dose scheduling of denileukin diftitox when used in combination with vaccine. By application of denileukin diftitox in melanoma patients, a recent study showed that denileukin diftitox depletes Treg cell in vivo significantly, resulting in enhanced immune functions and substantial development of antigen-specific CD8+ cells in peptide-vaccinated melanoma patients after immunization with respective MelanA/MART (melanocyte antigen recognized by T cells 1) and gp100 peptides (43). Most recently, it has been shown that Treg cell depletion improves endogenous anti-tumor immunity and the efficacy of active immunotherapy in animal models for cancer, suggesting that inhibiting Treg cell function could also improve the limited successes of human cancer immunotherapy. The strategies to block Treg activity have been identified. The fusion toxin denileukin diftitox reduces Treg umbers and function in the blood of some patients with cancer (30). Combining Treg cell depletion with active vaccination and other approaches pose additional challenges.
Because estrogen regulates Treg cell numbers in mice and promotes the proliferation of human Treg cells, Generali et al investigated the immunomodulatroy effects of aromatase inhibitor letrozole in breast cancer patients. They found that there is a significant reduction in Treg cells in letrozol and letrozole-"metronomic" cyclophosphamide patients after treatment (67). Importantly, this reduction in Treg cells is inversely related to the response. These novel findings suggest that letrozole has a significant immumomodulatory role and that aromatase inhibitors could be used in combination with other immunotherapeutic approaches. Recently, Powell et al have evaluated the Treg cell depleting capacity of the CD25-directed immunotoxin, RFT-SMPT-dgA and demonstrated that administration of RFT5-SMPT-dgA to patients with metastatic melanoma induced a transient but robust reduction in number of CD25highCD4 T cells in vivo (172). Using a highly metastatic breast cancer model, a most recent study showed that lung metastasis required CCR4+ Treg cells. Thus strategies that abrogate any part of this process should improve the outcome through activation of effector cells and prevention of tumor cell migration. They prevented lung metastasis by killing CCR4+ cells through delivery of TARC-fused toxin or depleting Treg cells (173).
Recently, using a syngeneic intracranial glioblastoma mouse model, Curtin et al demonstrated that systemic depletion of Treg cells 15 days after tumor implantation using CD25 depleting antibodies (PC61) resulted in a decrease in Treg cells present in tumor, draining lymph nodes and spleen and improved long-term survival. No improvement in survival was observed when 24 days after tumor implantation, suggesting that tumor burden is an important factor for determining efficacy of Treg depletion in clinical trials (174). They noted that this approach will be useful in a setting of minimal residual disease. Therapeutic modulation of Treg cell populations is clinically feasible and will potentially allow the induction of effective antitumor immune responses when combined with currently available immunotherapeutic strategies (35). Removal of Treg cells via a Bax-dependent apoptotic pathway significantly enhances anti-self antigen immune response, which demonstrated the proof of principle that apoptosis of Treg cells is a new therapeutic target (175). Therefore, continued characterization of Treg cell apoptosis and homeostasis pathway would further improve our understanding in regulation of immune response by cytokines, hormones and drugs, and would also lead to development of new therapeutics. Selective separation of functional CD4+CD25+ Treg cells from large-scale samples (patient leukapheresis samples) can be performed in large scale under clinical-grade conditions with sufficient selection, recovery, viability, ability to expand, and function for potential use in adoptive immunotherapy (68).
As described previously, the STAT protein regulate many aspects of cell growth, survival and differentiation (133,176). Interestingly, constitutive STAT3 has emerged as one of the major molecule involved in the mechanisms governing cancer immune tolerance and inflammatory disease. STAT3 activation is frequently observed cancer patients. A direct role of for STAT3 in mediating the generation of suppressive functions in CD4+ T cells was raised by a study conducted in mice laking SOCS3, which is an inhibitor of STAT3 (133,176,177). In a series of elegant experiments, Pallandre et al showed that interfering with STAT3 signaling in vitro inhibits both iTreg cell conversion and suppressive functions of nTreg cells (133). In vivo, STAT3 knockdown decreases Treg cell generation. Furthermore, they showed that STAT3 knockdown enhances antitumor immune response. Therefore, it appears that STAT3 signaling play a major role in CD4+ T cell-mediated immune tolerance. Accordingly, their findings determine that STAT3 activation promotes Treg cells generation and that STAT3 is critical in the molecular pathway required for FoxP3 expression (133). The findings also convincingly suggest that STAT3 modulation should be taken into account when assessing how Treg cells contribute to inflammatory diseases and tumor immunosurveillance. The activation or inhibition of STAT3 may be targeted by pharmacological agents or microRNA strategies to promote efficient adaptive immune responses to restore antitumoral immune responses (133,177). Therefore, it appears that STAT3 signaling play a major role in CD4+ T cell-mediated immune tolerance. Evidently, the STAT3 signaling pathway regulates cancer metastasis and constitutes a potential preventive and therapeutic target for cancer metastasis (176). Recently, there was a report that activated STAT3 protein plays a critical role in the induction of breast tumor induced by STAT3 knockdown 4T1 cells in immunocompetent BALB/c mice by enhancing the expression of several important genes including c-Myc and the metastatic regulator Twist (177). These studies suggest that stable expression of siRNA for STAT3 has potential as a therapeutic strategy for breast cancer.
Interestingly, an early study showed that Treg cells selectively express Toll-like receptor (TLR)-4, -5, -7, and -8 (178). Exposure of Treg cells to the TLR-4 ligand lipopolysaccharide (LPS) induces up-regulation of several activation marker and enhances their survival/proliferation. Importantly, LPS treatment increases CD4+CD25+ cell suppressor efficiency by 10-fold (178). Peng et al described a mechanism linking TLR8 signaling to the control of Treg cell function, in which synthetic and natural ligands for human TLR8 can reverse Treg cell function (179). They also showed that adoptive transfer of TLR8 ligand-stimulated Treg cells into tumor-bearing mice enhanced antitumor immunity. Recently, new approaches to tumor immunotherapy and vaccination have focused on enhancement of effector T cell responses by targeting innate immune cells and the receptors that mediate their activation. Pathogen recognition receptors, in particular TTLRs have been the primary targets for activation of innate immune cells (178). Indeed, it has already been demonstrated that TLR signaling may directly regulate the suppressive function of Treg cells and that TLR agonists are potent adjuvants for infectious disease antigens and have also been used as immunotherapeutic agents in cancer patients (180). Adoptive transfer of TLR8 ligand-stimulated Treg cells into tumor-bearing mice enhanced antitumor immunity, suggesting TLR signaling could play a critical role in controlling immune response to cancer and other diseases (179). Synthetic oligodeoxynucleotides (ODNs) that contain immunostimulatory CpG (cytosine and guanine separated by a phosphate) motif (CpG ODN) trigger an immunomodulatory cascade (19). Jarnickie et al demonstrated that TLR ligands also promote the induction of IL-10 secreting Treg cells and a p38 inhibitor suppresses the generation of Treg cells and enhances the antitumor therapeutic efficacy of DCs pulsed with antigen and CpG (19). Furthermore, they reported that attenuating Treg cell induction by TLR agonists LPS, poly (I:C) or CpG through inhibition of p38 MAPK signaling in DCs enhances their efficacy as vaccine adjuvants and cancer immunotherapeutics, providing evidence that p38 is an important therapeutic target.
Radiofrequency thermal ablation (RFTA) is a minimally invasive technique that has prove to be effective in the treatment of nonsurgical patients with liver cancer (28). This treatment destroys tumor tissue by delivering a high-frequency alternating current with ionic agitation and frictional heating. As a consequency, cell membrane alteration, protein denaturation, and necrosis around electrode are produced and large amounts of tumor and tissue debris are released (28). Of great interest, using this RTTA, in a series of elegant studies, Fietta et al assessed whether RFTA could affect multiple systemic inflammatory and immunological parameters, including CD25+FoxP3+ Treg cells in patients with primary or metastatic lung tumors (28). They demonstrated that 3 days after RFTA, a moderate and temporary systemic inflammatory response developed. Moreover, 30 days after RFTA, a significant reduction in CD25+FoxP3+ cell counts with an increase in CD4+ T-cell proliferation was observed. The reduction in CD25+FoxP3+ cells lasted up to 90 days after treatment. The use of RFTA in lung cancer patients has an immunomodulatory activity. They also reported that in addition to tumor ablation, downmodulation of Treg cells might be an important mechanism involved in the long-term clinical efficacy of RFTA. Their findings suggest that the small but significant reduction of the peripheral CD25+FoxP3+ Treg cell pool might be of functional importance in improving host T-cell reactivity. Further analysis on a large series of cancer patients will be required to carry out to corroborate these preliminary results.
THERAPEUTIC VACCINATION
Prophylactic cancer vaccines have been effective in a number of spontaneous tumor models in mice, including models of breast, prostate, and colon cancer (3). In cancer patients, vaccination strategies have been limited to advanced disease and many strategies for generating therapeutic immune response to cancer have been attempted (3). An early study showed that significant increases in the number of antigen-specific CTL in circulation after immunization of melanoma patients with peptides or melanoma tumor lysate-loaded with APC-based vaccines (181). A recent study showed that vaccination of mice with FoxP3 mRNA-transfected DCs elicited a robust FoxP3-specific CTL response and potentiated vaccine-induced protective immunity (182). Importantly, FoxP3 vaccination led to the preferential depletion of FoxP3-expressing Treg cells in the tumor but not in the periphery, whereas αCD25 antibody treatment led to depletion of Treg cells in both the tumor and the periphery. These studies strongly indicate that depletion of Treg cells by treatment with αCD25 antibody may synergize with vaccination protocols to engender protective immunity in mice. A recent study elegantly showed that breast cancer patients have a greater percentage of circulating Treg cells along with serum levels of TGFβ than normal subjects, and that HER2/neu peptide (E75) vaccination increased circulating activated CD4+ T cells post-vaccination (147). However, Treg cells were significantly reduced after vaccination. Importantly, post-vaccination decreases in Treg cells were temporally associated with increased E75 vaccine-specific CD8+ T cells and corresponding HER/neu+ tumor cytotoxicity. These studies suggest that successful cancer vaccination strategies may require the alteration of complex immune interaction. A more recent study reported that melanoma patients receiving a MAGE-A3.DP4 peptide vaccine can produce CD4+ T cells that may exert Treg cell function in vivo (183). NY-ESO-1 is a 180 amino-acid human tumor antigen expressed by "cancer-testis" antigen (20). In human, NY-ESO-1 is a highly immunogenic tumor antigen expressed in a variety of malignancies, making it an excellent target for cancer vaccination (20). Nicholaou et al recently developed a vaccine consisting of full-length recombinant NY-ESO-1 protein formulated with ISCOMATRIX adjuvant, which generated humoral and T-cell-mediated immune responses and seemed to reduce the risk of disease relapse in patients with fully resected melanoma (20). They examined the clinical and immunologic efficacy in patients with advanced metastatic melanoma. They found that in contrast to minimal residual disease, advanced melanoma patients showed no clinical responses to vaccination. Although strong antibody responses were mounted, the generation of delayed-type hypersensitivity response was significantly impaired and patients with advanced melanoma had a significantly higher proportion of circulating CD4+CD25+FoxP3+ Treg cells compared with those with minimal residual disease. Their results point to a tumor-induced systemic immune suppression, showing a clear association between the stage of melanoma progression, the number of Treg cells in the blood, and the clinical and immunologic efficacy of the NY-ESO-1SCOMATRIX cancer vaccine (20). Interestingly, a most recent study demonstrated that only Treg cell depletion followed by neu DNA vaccination abrogated tolerance to neu (rat erbB-2), resulting in complete regression of neu+ tumors, as well as long-term protection from spontaneous tumorigenesis (184). In addition, in mice with induced tumor regression, mouse thyroglobulin response was enhanced with modest increases in autoimmune thyroiditis. These studies strongly suggest that tumor regression induced by Treg cell depletion and DNA vaccination can exacerbate autoimmunity. Therefore, patients undergoing immunotherapy should be closely monitored for signs of autoimmune symptoms.
PROGNOSTIC VALUE OF Treg CELLS IN PATIENTS WITH CANCER
A better prognosis was observed in patients with a low prevalence of Treg cells and a high prevalence of Treg cells seems to be a marker of poor prognosis in pancreatic ductal carcinoma (185). A recent study also showed that in patients with undesirable outcome the balance is tipped in favor of Treg cells, whereas in patients with relatively desirable outcome, the balance is tipped toward effector T cells, suggesting that Treg cells are associated with hepatocellular carcinoma invasiveness, and intratumoral balance of regulatory and CTL is a promising independent predictor for recurrence and survival in hepatocellular carcinoma (124). A combination of depletion of Treg cells and concomitant stimulation of effector T cells may be an effective immunotherapy to reduce recurrence and prolong survival after surgery. The patients with high expression of CD4 or CD8 had distinctly worse cancer specific overall survival (38). The analysis of tumor-infiltrating immune cells may be a valuable prognostic tool and a marker of lymph node involvement. Shevach reported that an elevation in the number of tumor Treg cells was a significant predictor of patients death, even after controlling for stage of disease and surgical reduction of tumor mass (186). In addition, one line of evidence showed that the presence of FoxP3+ cells within tumors has been shown to be predict the prognosis, invasiveness, and metastatic ability of some tumors by modulating the ability of the immune system to target tumor cells. The widespread use of FoxP3 as a biomarker should be explored for human tumors to enable physicians to make better decisions in oncologic care and to prepare the filed of novel therapeutic agents directed at the elimination of Treg cells within tumors (39). FoxP3 expression might be related to the metastatic potential of the tumor rather than to suppression of a specific immune response (187). The findings that FoxP3 can be expressed by not only tumor-infiltrating Treg cells but also tumor cells has two important implications (187). First, increased levels of FoxP3 mRNA expression may be a result of not only an increased influx of Treg cells but also the increase expression of FoxP3 directly in tumor cells. Second, we need to recognize that FoxP3-targeted therapy may need to be targeted at not only Treg cells but also FoxP3-positive tumor cells. The expression of FoxP3 in tumor cells indicates that FoxP3-targeted drugs must to be able to penetrate into the tumor, which is much more challenging than depleting FoxP3 in the periphery (187). FoxP3 remains an accurate marker to define primary Treg cells in patients with cancer and autoimmune disease (188). The authors suggest that the combination of FoxP3 and cytokine profile is useful for further functionally distinguishing primary Treg cells from activated conventional T cells. Most recently, a study demonstrated that FoxP3 expression in breast cancer was associated with worse overall survival probability and the risk increased with increasing FoxP3 immunostaining intensity (148). FoxP3 was also a strong prognostic factor for distant metastases-free survival. The probability of 10-year survival in node-negative subgroup was 100% for FoxP3-negative and 82% for FoxP3-positive patients. Authors proposed that FoxP3 expression as a new independent prognostic factor in breast carcinoma, which might help to improve the selection of patients for appropriate therapy (148). In malignancies of intrinsic central nervous system, interestingly, Heimberger et al have recently shown that Treg infiltration differed significantly in the tumors according to lineage, pathology, and grade, and that Treg cells seemed to have the highest predilection for tumors of the astrocytic lineage and specifically in the high-grade gliomas, such as glioblastoma multiforme (41). Intratumoaral FoxP3-positive regulatory immune cells significantly increased during IL-2-based immunotherapy, and high numbers of on-treatment FoxP3-positive cells were correlated with poor prognosis in patients with metastatic renal cell carcinoma (139).
In marked contrast to the poor prognostic value of Treg cell presence in these tumors, some current studies demonstrated that disease specific survival was not influenced by the presence of FoxP3+ Treg cells in ovarian tissue, but median disease specific survival of patients with a high CD8+/FoxP3+ ratio in ovarian-derived tumor tissue was twice as high as for patients with a low CD8+/FoxP3+ ratio (141). Possibly, the positive prognostic effect of the of FoxP3+ cells in their samples can be attributed to the staining of not only suppressive, but also activated T cells expressing FoxP3. Furthermore, a widely used model for human colorectal carcinogenesis is the multiple intestinal neoplasia (Min) mouse, which has a germ-line mutation in the Apc tumor suppressor gene (ApcMin). Interestingly, a recent study has shown that adoptive immunotherapy using CD4+CD25+ Treg cells prevents the development of intestinal tumors and rapidly induces regression of established tumors in the ApcMin/+mouse model of human intestinal cancer (185). Surprisingly, a study reported that FoxP3+ Treg cell density in normal and tumor tissue had stronger prognostic significance in colorectal cancer compared with CD8+ and CD45RO+ lymphocytes. The authors reported better survival associated with a high density of intratumoral FoxP3+ Treg cells in colorectal cancer (26). In patients with follicular lymphoma and Hodgkin's lymphoma, it is now well established that high number of intratumoral Treg cells is associated with longer disease-free and overall survival. This correlation between the number of Treg cells and favorable clinical outcome has also recently been extended in some solid tumors (189). In patients head and neck cancer, the authors found that tumor infiltration by FoxP3+CD4+ Treg cells was positively associated with better locoregional control of the tumor. In addition, multivariate analysis showed that the only significant prognostic factors related to locoregional control was T stage and Treg cell infiltration of the tumor (190). Frequency of FoxP3+CD4+ Treg cells has been shown to be higher in patients with no evidence of disease after oncologic therapy than in patients with active disease (123). These results were surprising, and in marked contrast to the poor prognostic value of Treg cell presence in other tumors as described previously (40,91,125,139,148,190,191). Interestingly, a recent study showed that it is the Foxp3- subset of CD4+CD25+ T cells and not the FoxP3+ subset that correlated with worse pathologic feature of renal cell carcinoma and cancer-specific survival (40). Various parameters may explain these seemingly contradictory results regarding prognostic value of Treg cells in patients with cancer. First, the role of Treg cells seems to differ according to tumor stage (192). Second, it is not surprising that various studies have reported that the ratio of CD8+ T cell to Treg cells had more important on patient survival than the number of Treg cells or intraepithelial CD8+ T cells alone (124). Third, unlike in mice, FoxP3 expression is not confined to certain population in humans (187).
A VIEW TO THE FUTURE
The pathophysiological role of tumor-infiltrating CD4+ Treg cells remains less understood than that of CD4+ Treg cells. However, recently, a study showed that the conversion of CD8+ effector T cells into suppressor cells in tumor microenviroment and tumor restrict the immune response from effectively controlling tumor growth (193). As subsequent effectors infiltrate the tumor, the tumor-induced Treg cells may suppress these new effectors and reduced their ability to confer tumor immunity. This cyclic suppressive process may contribute to the profound loss of antitumor response following immunotherapy. New approachs to block these pathway will enhance our ability to generate more sustained and effective antitumor T cell response (193). Most recently, one study showed that blocking TGFβ in a tumor-bearing mice can inhibit the induction of highly suppressive tumor-sensitized Treg cells, indicating that TGFβ blockade may improve the generation of therapeutic immune responses in patients with cancer (194). As already outlined, FoxP3 is a key transcription factor for development and function of nTreg cells. Most recently, Sakaguchi group reported that human FoxP3+CD4+ T cells were composed of three phenotypically and functionally distinct subpopulations (195): CD45RA+FoxP3lo resting Treg cells (rTreg cells) and CD45RA-FoxP3high activated Treg cells (aTreg cells) and cytokine-secreting CD45RA-FoxP3lo nonsuppressive T cells. They noted that the dissection of FoxP3+ cells into subsets enables one to analyze Treg cell differentiation dynamics and interactions in normal and disease states, and to control immune response through manipulating particular FoxP3 subpopulations. Currently, one study showed that MHC class II expression identifies an effector subset of human CD4+CD25high FoxP3high natural T cells (DR+ Treg cells) that induces more rapid suppression and exhibits higher FoxP3 expression than the remaining Treg populations (196). This paper reported that responder cells actively kill effector Treg cells producing granzyme B in response to strong TCR stimulation. These data demonstrate a novel role for granzyme B to down-regulate effector DR+ Treg cell function.
Intriguingly, multiple investigators have shown that microRNA (miRNA)-mediated RNA interference has been identified as a novel mechanisms that regulates protein expression at the transcription level (197-199). Furthermore, recent publications have provided compelling evidence that a range of miRNAs are involved in the regulation of immune responses, including the development and differentiation of B and T cells, especially Treg cells (198-200). In the absence of miRNAs, FoxP3+ Treg cell develop but fail to maintain immune homeostasis, leading to a scurfy-like disease. In addition, miRNA-155 deficient mice have reduced numbers of Treg cells, both in thymus and periphery (200,201). A most recent study showed that FoxP3 controlled the elevated miR155 expression required for maintaining Treg cell proliferative activity and numbers under non-lymphopenic conditions, suggesting FoxP3-dependent regulation of miR155 maintain competitive fitness of Treg cell subset by targeting SOCS1 protein, a negative regulatory of the IL-2 signaling cascade (197). Overall, there is now compelling evidence that miRNAs are involved in the regulation of the immune response, and modulation of their activity might ultimately provide a novel therapeutic approach in the treatment of inflammatory disease and certain cancers (197,200,202).
The ability to overcome some of the limitations of Treg cell therapy by genetically re-programming Treg cells is potentially attractive. Lastly, as our knowledge of Treg cell immune-suppressive mechanisms increases, additional opportunities to enhance or inhibit Treg cell function via gene therapy will be encouraging. It is becoming increasingly clear that a major component of the next wave of therapeutic agents that will attempt to tackle our unmet medical needs will be Treg cell and gene therapy. Indeed, Treg cell therapy has the promise of avoiding many of the toxicities observed with current drug regimens. This potential will be realized too early.
CONCLUSION
It is now absolutely clear that there now exists overwhelming evidence from both human and animal studies that regulatory T cells play an active and significant role in the progression of cancer, and that regulatory T cells have an important role in suppressing tumor-specific immunity. Thus, there is a clear rationale for developing clinical strategies to diminish there regulatory influences, with the ultimate goal of augmenting antitimor immunity. Therefore, manipulation of Treg cells, including depletion, blocking trafficking into tumors, or reducing their differentiation and suppressive mechanisms, represent new strategies for cancer treatment. It should be mentioned that although this review has focused on one of the most well characterized regulatory T cell types, many other subsets including Tγ1, Th3, CD8+ regulatory cells, CD4+ cells that show features of both natural and adaptive Treg cells, and γδ T cells have been reported. Thus, future challenge include the identification of unique regulatory cell markers that can be used to more specifically target these cells in vivo and a more accurate characterization of the molecular nature of immune suppression. Therapeutically, agents that can selectively and efficiently target Treg cells deserve extensive research. Survival is the most important end-point for evaluating cancer therapy. For the last 50 years cytotoxic chemotherapy has been one of the mainstay of cancer treatment. While the ability of chemotherapy treatment to induce tumor control and remission has continued to improve (notably in the hematological malignancies), chemotherapy often fails to prevent disease recurrence. Successful combination of chemotherapy plus radiotherapy with immunotherapy would appear to be the best way forward for improving the ability of chemotherapy-based treatments to prevent disease recurrence and increase cure rates of many malignant diseases. Although targeting Treg cells is an attractive option in treating human tumors and "all anyone is talking about these days is regulatory T cells", it is probable that a complicated combinatorial therapeutic regimen of traditional tumor therapy and conventional immunotherapy combined with novel tumor immunotherapy, including targeting Treg cell (chemoradiosurgicoimmunotherapy) would appear to be the best way forward to obtain effective, reliable and consistent clinical efficacy. Researchers, clinical immunologists and oncologists, together, should make efforts to design combined chemoradioimmunotherapy strategies (see Fig. 2). However, there are still many unanswered questions. While the road is not straight and smooth, there are challenge and great promise and excitement over the horizon. It will address both challenge and opportunities in human Treg cell therapy. Only time and hard work will answer many questions we are now confronting.

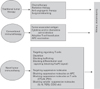

 XML Download
XML Download