

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Leukocyte-endothelial Interactions
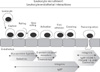
Leukocytes circulating in the blood need to migrate into tissues where a pathogen- or nonpathogen-derived antigen or inflammation is present. In particular, leukocyte recruitment is pivotal during infection caused by bacterial, viral, fungal or parasitic pathogens, during inflammatory disorders, and in the course of autoimmune diseases. All leukocytes participating in innate or adaptive immunity have the ability to migrate to the sites of inflammation or tissue injury by crossing endothelial barriers between blood and tissues (1,2). The process of leukocyte recruitment comprises a tightly regulated cascade of adhesive interactions between leukocytes and endothelial cells; (i) leukocyte capture by activated endothelium, (ii) leukocyte rolling on the endothelium, (iii) slow rolling of leukocytes on the endothelium, (iv) leukocyte activation by chemokines on the endothelium, (v) firm adhesion of leukocytes onto the endothelium, (vi) post-adhesion leukocyte crawling or locomotion, and (vii) transendothelial migration or diapedesis. Distinct families of adhesion molecules control which cells are to be correctly recruited to the right place at the right time (Fig. 1) (3,4).
ADHESION MOLECULES THAT REGULATE LEUKOCYTEENDOTHELIAL INTERACTIONS
At sites of inflammation, the endothelium is locally activated by cytokines to express adhesion molecules called selectins on its surface where the leukocytes are captured from the blood and start rolling along the endothelial surface, which is the initial step of the leukocyte-endothelial interactions. Capturing and rolling are primarily mediated by the interaction between endothelial selectins and their glycosylated leukocyte selectin ligands. Under certain circumstances, however, capturing and rolling are mediated by the interaction between leukocyte integrins α4β1 (VLA-4) and α4β7 and their endothelial ligands VCAM-1 and MadCAM-1, both of which are members of immunoglobulin superfamily (5-9).
The leukocyte rolling on the vessel wall allows the interaction of G-protein coupled receptors (GPCRs) on the leukocyte surface with the specific chemokines which are clustered and immobilized via binding to glycosaminoglycans in the endothelial glycocalyx, thereby priming the GPCRs (10-12). Activation of the GPCRs triggers an inside-out signaling cascade which induces integrin clustering at contact site and rapid conformational change in integrins to an active state. This allows efficient binding of leukocyte β1 and β2 integrins, i.e., LFA-1, Mac-1 or VLA-4, to immunoglobulin superfamily receptors, ICAM-1, -2 or VCAM-1, on the endothelium, resulting in leukocyte arrest and firm adhesion to the vessel wall (13,14). Selectin-mediated initial leukocyte rolling and subsequent integrin activation may also cooperate to mediate an additional step termed slow rolling prior to the step of firm adhesion (8,15).
Once firmly adhered, leukocytes crawl or locomote along the endothelial surface to find a junction between two endothelial cells (paracellular route) or to find a way to pass through one endothelial cell (transcellular route). Formation of transmigratory cups by leukocyte integrin LFA-1 and its endothelial ligand ICAM-1 is essential in both paracellular and transcellular routes (16-19). Ligation of ICAM-1 by leukocyte integrins, followed by intracellular signaling events, opens up endothelial junctions (20-23). Transmigration of leukocytes through the endothelial layer is achieved by a chain of adhesive events which are orchestrated by a number of adhesion molecules. Several homophilic and heterophilic interactions between the leukocyte/endothelial apical and junctional adhesion molecules take place sequentially (24-26). Molecules directly acting in the transmigration include leukocyte integrins, endothelial immunoglobulin superfamily members (ICAM-1, -2, JAM-A, -B, -C and PECAM-1) and a non-immunoglobulin molecule CD99 (27-31).
The aforementioned classical leukocyte-endothelial interactions in the leukocyte recruitment do not occur in all tissues. Non-classical leukocyte endothelial interactions are regulated by the tissue-specific microvasculature environment with unusual combinations of distinct endothelial adhesion molecules and chemokines, and tissue-specific signaling pathways (32-35). For example, leukocyte migration to the liver primarily takes place in the hepatic sinusoids, not in the post-capillary venules as is the case with many other tissues. As the hepatic sinusoids have non-classical endothelial adhesion molecules and a set of unique predominant chemokines on the vascular endothelium, unusual leukocyte adhesion molecules are employed (33,35-37).
Although immune cell migration is critical for protective immune responses against pathogens, the accumulation of leukocytes in the tissues could trigger significant release of cytotoxic mediators from leukocytes, leading to tissue damage and finally a wide spectrum of inflammatory conditions. Thus, inhibition of excessive or misdirected leukocyte recruitment provides a means for anti-inflammatory therapeutics.
DEVELOPMENTAL ENDOTHELIAL LOCUS-1 (DEL-1): STRUCTURE AND EXPRESSION
Del-1 (also termed Edil3) is a 52 kDa glycoprotein secreted by endothelial cells and associates with the endothelial cell surface and extracellular matrix, but seems not to circulate in the blood (38-40) (Choi unpublished). Human Del-1 shares more than 97% amino acid identity with the mouse counterpart. Del-1 consists of a signal sequence at its N-terminus, three epidermal growth factor (EGF)-like repeats and two discoidin I-like domains at its C-terminus (Fig. 2) (40). It has been shown that the discoidin domains are responsible for binding to sulfated glycan and phosphatidyl serine-rich membrane (39,40). On the other hand, the second EGF like repeat contains an Arg-Gly-Asp (RGD) motif that enables Del-1 to bind integrin αvβ3, mediating adhesion of endothelial cells to Del-1 (40).
Expression of Del-1 was initially observed in embryonic cells including endothelium and thymus, and subsequent studies showed its expression in adult endothelial cells, some subsets of macrophages and hypertrophic chondrocytes, It was recently reported, however, that Del-1 is expressed in adult mice in a tissue-specific manner: strong expression in the lung, brain and eye, a very little in the kidney, and none in the intestine, liver, heart, spleen, whole blood and bone marrow cells (38).
INHIBITION OF LEUKOCYTE-ENDOTHELIAL INTERACTION BY DEL-1
A role for Del-1 may be inferred from its expression in adult vascular endothelium. Recently, it was demonstrated that Del-1 functions as an endogenous inhibitor of a major leukocyte adhesion receptor LFA-1 to prevent leukocyte adhesion to the endothelium (38). Unlike many adhesion molecules including selectins and Ig superfamily members promoting leukocyte adhesion on the endothelium, Del-1 inhibits the process of leukocyte binding to the endothelium, thereby suppressing entrance of leukocytes to inflamed tissues.
Del-1's loss of function phenotype is characterized by an increase in leukocyte adhesion on the vascular endothelium and increased inflammatory cell recruitment. Del-1 deficient mice showed more leukocytes adhering on the post-capillary vasculature under both non-inflamed and inflamed conditions, than wild type mice, as demonstrated by intravital microscopy (38). Increased adhesion of leukocytes to the vascular endothelium was a prelude to an increase in leukocyte migration into the inflamed tissue. Intranasal administration of LPS to the Del-1 deficient mice to induce lung inflammation yielded more neutrophil accumulation in bronchoalveolar lavage fluid, as opposed to control mice (38). This phenotype was reversed by the absence of LFA-1 on leukocytes. Upon LPS-elicited lung inflammation, LFA-1/Del-1 double deficient mice display very low leukocyte recruitment, indicating that the phenotype of Del-1 deficiency is dependent on the presence of LFA-1 (38).
Given that Del-1 is a ligand for LFA-1, it is intriguing that Del-1 inhibits the function of LFA-1 (38). Other LFA-1 ligands including ICAM-1 are known to enhance leukocyte adhesion. However, Del-1 is an exception. Under physiologic flow conditions, addition of Del-1 to the system where leukocytes adhere onto immobilized ICAM-1 via interaction with activated LFA-1 represses the adhesion of leukocytes (38). This implies that Del-1 may compete with ICAM-1 for the binding to LFA-1. Studies on the mechanism by which Del-1 inhibits LFA-1 functions are underway.
Del-1-mediated inhibition of the interaction between LFA-1 and ICAM-1 further implies that Del-1 could be used as a therapeutic tool for inflammatory diseases. In such a case, a fair amount of exogenous Del-1 is worth to employ, given that ICAM-1 is up-regulated in the tissues during the course of inflammation. Following LPS-elicited lung inflammation, Del-1 deficient mice supplemented with soluble Del-1 behaved in a comparable fashion to the wild type mice in terms of inflammatory cell recruitment into the lung tissues, recapitulating Del-1's potential as anti-inflammatory therapeutics (38).
Modulation of Del-1 expression could also represent a candidate therapeutic strategy for autoimmune and chronic inflammatory disease. Del-1 mRNA message was down-regulated in the inflamed tissues and the endothelial cells upon induction of inflammation by TNF-α stimulation (38). It is reasonable therefore that as a resident anti-inflammatory molecule, such a decrease in Del-1 expression may render the body's normal inflammatory response more competent. It is tempting to speculate that constitutively expressed Del-1 in certain tissues may function as a gate keeper within the tissues against unspecified potential weak inflammatory agents. As a result, normal healthy subjects should not respond to the agents, thereby preventing the initiation of immune response. However, those who have defects in the expression and/or function of Del-1 may show hypersensitivity. Taken together, Del-1 holds promise for treating diseases related to excessive or deficient recruitment of leukocytes. If excessive recruitment is required (i.e., infectious disease), a strategy to inhibit Del-1 should be applied. Conversely, if excessive leukocyte is the culprit, a strategy to boost Del-1 should be applied.

 XML Download
XML Download