

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
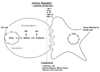
NK cells are large granular lymphocytes involved in protection against infectious microbial pathogens and tumors. They are found throughout the body in both non-lymphoid peripheral tissues and lymphoid organs. Unlike B and T lymphocytes which express somatically rearranged antigen-specific receptors, NK cells express receptors to recognize and respond to an array of infected or tumorigenic cells (1). Decades of work have resulted in a substantial gain in understanding of what and how NK cells are regulated before they kill target cells, lending important insights into their gene regulation mechanisms for immune regulation and cytotoxicity (Fig. 1).
NK cells actively respond to environmental changes by regulating de novo gene expression, which is strictly controlled by four-step-expression. Four-step-expression is synthesis and degradation of mRNA together with synthesis and degradation of the corresponding proteins (2). Fine tuning of synthesis and degradation rates is not only essential for maintaining protein levels, but also allows for fast and sensitive responses to target cells. In contrast to B and T cells, the post-transcriptional mechanisms governing NK cell activation remain poorly understood. However, there are some examples of post-transcriptional gene regulation during the trafficking, immune synapse formation, cytokine production, and cytolysis steps of NK cell responses (Table I). This review focuses on possible regulatory mechanisms of mRNA stability and translation during NK cell activation.
Urokinase plasminogen activator (uPA) and uPA receptor (uPAR)
The uPA system has been shown to play a major role in the extravasation and migration of leukocytes into areas of inflammation. One unique feature of the uPA system is the presence of a specific cellular receptor for uPA, the uPA receptor (uPAR), which is involved in cell migration and invasion, independent of its role in the proteolytic pathways. uPAR has been shown to be involved in cellular adhesion by its ability to bind to the extracellular matrix protein vitronectin. uPAR also has the capacity to initiate secondary signaling pathways through its interactions with integrins, thereby promoting cellular movement and migration (3).
The regulation of uPA and uPAR expression occurs at a post-transcriptional level through mRNABPs that bind to the 3'-UTR. Heterogeneous nuclear ribonuclear protein C1 (hnRNP C1) and HuR bind to the uPA 3'-UTR, which leads to uPA mRNA stabilization (4). Similarly, hnRNP C1 binds to a 110-nucleotide sequence of the uPAR mRNA 3'-UTR, thereby preventing its degradation (5). Conversely, p53 decreased both uPA and uPAR mRNA stability by binding to a 35-nucleotide sequence in the uPA 3'-UTR and a 37-nucleotide sequence in the uPAR 3'-UTR, resulting in decreased cellular expression. p53-deficient (p53-/-) lung carcinoma cells express robust levels of cell surface uPA and uPAR mRNA, thereby contributing to human airway epithelial or lung carcinoma cell viability (6,7).
A recent study showed that NK cells expressed both uPA and uPAR and that in vitro NK cells employ the uPA system following stimulation with IL-2. uPA and uPAR mRNA binding proteins (mRNABPs) were also detected in NK cells. Increases in uPA and uPAR following IL-2 stimulation, correspond to changes in uPA and uPAR mRNA-mRNABP interactions. The upregulation of uPA and uPAR may partially explain the increased NK cell invasiveness following IL-2 treatment (8).
NKG2D ligands
An encounter between an immune cell and another host cell may result in the generation of an immune synapse, a specialized interface at the cell-cell contact point. Immunological synapses (IS), as described by Davis, are contacts between two cells, at least one of them being a cell of the immune system, such as NK cells, that results in segregation of proteins at the cell-cell interface into micrometer-scale three-dimensional domains (9). NK cell activation is controlled by a dynamic balance between stimulatory and inhibitory pathways that are initiated upon interactions with potential target cells (1). NKG2D is one of the best characterized activating receptors found on NK and CD8+ T cells. This receptor recognizes several different ligands (MHC class 1 polypeptide-related sequence A/b [MICA/B] and UL16-binding proteins [ULBPs]) induced by cellular stress and infection. NKG2D-ligand engagement activates NK cells and drives cytotoxicity against the target cells (9).
MICA and MICB are stress-induced ligands recognized by the activating receptor NKG2D. Recently, Mandelboim and colleagues reported MICA/B-targeting of viral and cellular microRNAs (miRNAs). The miRNA hcmv-miR-UL112 encoded by human cytomegalovirus down-regulates MICB expression by targeting a specific site in the MICA and MICB 3'-UTRs during viral infection. The down-regulation of MICB leads to decreased binding of NKG2D and reduced killing by NK cells. Notably, this is a novel viral immunoevasion mechanism based on miRNA regulation (10). Furthermore, a group of endogenous cellular miRNAs (miR-106b, -20a, -373, -520d, and -93) that bound to the MICA and MICB 3'-UTR sequences was identified. These miRNAs share the mRNA target site with the viral miRNA hcmv-miR-UL112. These miRNAs repress MICA/B under normal conditions to maintain their expression under a certain threshold and they facilitate acute upregulation of MICA/B during cellular stress (11).
Granzyme B and perforin
When a fully primed NK cell recognizes a target cell, an IS is formed between the two cells. Cytotoxic granules within the NK cells move toward the synapse, fuse with the plasma membrane, and release their contents into the synaptic cleft. Perforin then facilitates the delivery of granzymes into the cytosol of the target cell, where a variety of substrates are cleaved to initiate cell death (12). Serine protease granzymes are key NK cell effector molecules, but their regulation remains largely undefined. Granzyme B, the most thoroughly characterized of the granzymes, cleaves a variety of procaspases, BID, inhibitor of caspase-activated DNase, and other intracellular substrates to initiate classical apoptotic pathways (13).
Resting NK cells are minimally cytotoxic against tumor target cells in vitro. Cytokine-induced in vitro activation of NK cells results in potent killing associated with a dramatic increase in granzyme B and perforin, with minimal changes in corresponding mRNA abundance. This suggests that resting NK cells are "pre-armed" with high amounts of granzyme B and perforin mRNA. Both mRNAs are repressed during normal mRNA translation, but are released by cellular activation [14]. Furthermore, it is thought that this unknown mechanism is an efficient arming process for cytolytic immune cells, and allows for fast and sensitive responses to target cells during NK cell activation.
Interferon-γ
IFN-γ has been extensively studied and has been found to profoundly affect a variety of immune responses. These include upregulation of major histocompatibility complex class I and II expression, proliferation and differentiation of lymphocyte populations, and induction of genes that code for immunomodulatory proteins, such as tumor necrosis factor alpha (TNFα) and nitric oxide synthase. T and NK cells are the primary cellular producers of IFN-γ. In NK cells, IFN-γ production is triggered by interactions with target cells, such as tumor or virally infected cells, or by a variety of cytokines, such as interleukin-2 (IL-2) and IL-12. These cytokines can act independently to induce IFN-γ expression. However, together they act synergistically to induce large amounts of IFN-γ. The kinetics of IFN-γ expression results from both transcription and rapid degradation of IFN-γ mRNA (15).
As with many cytokine mRNAs, IFN-γ mRNA contains an AU-rich element (ARE) in its 3'-UTR. AREs have been identified in the 3'-UTR of a variety of labile mRNAs, and are well known to lead to decreases in the half-life of corresponding target genes. ARE-binding proteins as trans-acting factors include members of the Hu protein family HuR, hnRNP D (AUF1), tristetraprolin (TTP), the K homology-type splicing regulatory protein (KSRP), the T-cell restricted intracellular antigen (TIA)-1, and TIA-related protein (TIAR) (16). IFN-γ mRNA degradation is mediated by TTP and a 70-nucleotide AU-rich sequence in its 3'-UTR. TTP knock-out mice showed overexpression of IFN-γ due to stabilization of the IFN-γ mRNA, suggesting that TTP plays an important role in turning off IFN-γ expression at the appropriated time during an immune response (17,18). Likewise, IFN-α and -β mRNAs contain AREs and are subject to mRNA destabilization (19,20).
Granulocyte-macrophage colony-stimulating factor (GM-CSF)
Granulocyte-macrophage colony-stimulating factor (GM-CSF) is a multifunctional cytokine currently used for the reversal of neutropenia associated with bone marrow and haemopoietic stem cell transplantation and chemotherapy. GM-CSF also modulates the function of differentiated white blood cells. In local inflammatory responses, GM-CSF stimulates antimicrobial and antitumor effects of macrophages. GM-CSF further enhances healing by its actions on fibroblasts and epidermal cells. GM-CSF may enhance antibody dependent cellular cytotoxicity (ADCC) in several cell types and cytotoxicity of NK cells. GM-CSF may be useful for inducing or augmenting antibody responses to antimicrobial vaccines, to enhance killing of intracellular microorganisms, to accelerate epidermal and mucosal wound healing, and to stimulate protective immunity against tumors (21). In multiple murine models, GM-CSF proved to be the most potent immunostimulatory molecule (22).
Degradation of the ARE-containing GM-CSF mRNA is accelerated in vitro by protein fractions enriched for hnRNP D, an ARE-specific binding factor (23). hnRNP D is broadly involved in mRNA decay (24,25). HuR, a ubiquitously expressed member of the Elav family of RNA binding proteins, exhibits specific affinities for ARE-containing RNA sequences in vitro, which correlates with their in vivo decay rates. Overexpression of HuR enhances the stability of GM-CSF mRNA (26). Enzymes involved in all three of these mRNA decay processes, as well as 5'-to-3' exonucleolytic decay, include the protein TTP and its homolog BRF-1, which bind to GM-CSF ARE and activate mRNA decay. The TTP family functions as a molecular link between ARE-containing mRNAs and mRNA decay machinery by recruiting mRNA decay enzymes. The TTP protein family also helps to explain how deadenylation, decapping, and exonucleolytic decay can be independently activated on ARE-containing mRNAs (27,28).
Interestingly, the GM-CSF ARE has also been shown to control translation in vitro. In the absence of a native 5'-UTR, the ARE and poly (A) tail act in concert to block GM-CSF mRNA translation. Substitutions of different regions of the native 5'-UTR revealed that the entire sequence was essential in maintaining the highest rates of translation. The 5'-UTR is highly conserved, suggesting similar regulation in multiple species. The 5'-UTR is the dominant element regulating GM-CSF mRNA translation, overriding the inhibitory effects of the ARE and the poly (A) tail (29,30).
Interleukin-10 (IL-10)
IL-10 has been shown to be anti-inflammatory in many model systems. Deregulation of IL-10 leads to various immunological diseases, such as cancer, rheumatoid arthritis, asthma, and infectious disorders. Therefore, it is likely that IL-10 expression is tightly regulated at the transcriptional and posttranscriptional levels (31). Recently, it has been reported that NK cells are the major source of murine IL-10, as compared with helper T cells. The inhibitory effect of NK cells are only acquired later during infection, coincident with increased IL-10 mRNA stability and an enhanced capacity to secrete IL-10 protein. This suggests that post-transcriptional regulation of IL-10 mRNA is involved in the inhibitory function of NK cells. However, the detailed molecular mechanisms have yet to be elucidated (32).
The availability of IL-10 protein is significantly determined by post-transcriptional mechanisms. ARE in the 3'-UTR of IL-10 are involved in its translational regulation. Adenosine receptor ligands have anti-inflammatory effects and modulate immune responses by inducing IL-10 production by immunostimulated macrophages. Adenosine receptor activation acts by relieving the translational repressive effect of the IL-10 3'-UTR. Adenosine enhanced binding of proteins to a region of the IL-10 3'-UTR containing the GUAUUUAUU nonamer. During lipopolysaccharide (LPS) stimulation of THP-1 cells, the p40 hnRNP D isoform binds to the IL-10 3'-UTR and selectively induces IL-10 expression (33,34). Interaction of AREs in the 3'-UTR of IL-10 mRNA with hnRNP D leads to the degradation of IL-10 mRNA (35).
Moreover, a miRNA, hsa-miR-106a, that regulates IL-10 expression was reported. hsa-miR-106a directly binds to the IL-10 3'-UTR to repress translation. Also, the transcription factors Sp1 and Egr1 have an important role in hsa-miR-106a transcription and, thus, indirectly regulates the expression of IL-10 at the post-transcriptional level (36).
Tumor necrosis factor alpha (TNF-α)
TNF-α, initially discovered as a result of its antitumor activity, is a critical component of effective immune surveillance and is required for proper proliferation and function of NK cells, T cells, B cells, macrophages, and dendritic cells (37). Post-transcriptional regulation and stability is important for TNF-α expression, as TNF-α mRNA contains an ARE in the 3'-UTR. Regulation of TNF-α mRNA turnover has been shown to be mediated by the trans-acting proteins TTP and HuR, which bind the ARE and destabilize or stabilize the transcript, respectively (38-41). Among TNF-α ARE-binding proteins, TIA-1 and TIAR act as TNF-α mRNA translational silencers. In unstimulated immune cells, TNF-α mRNA is translationally repressed and becomes efficiently translated upon cellular activation (42,43).
Macrophage inflammatory protein-1α (MIP-1α)
MIP-1α, a chemotactic pro-inflammatory cytokine, is also secreted by NK cells. MIP-1α is a chemoattractant for monocytes and neutrophils and, thus, plays an important role in initiation and control of inflammation. In resting RAM cells, MIP-1α mRNA decayed rapidly with a half life of less than 2 hours. LPS treatment of RAM cells resulted in a dose-dependent increase in MIP-1α mRNA expression. The induction of MIP-1α mRNA by LPS was partially the result of mRNA stabilization, as half life increased to over 6 hours (44). Although the detailed molecular mechanism has not been defined, ARE-mediated mRNA degradation might be involved in the mRNA destabilization of MIP-1α, similar to those of other cytokine mRNAs.
CONCLUSION
Each step of NK cell activation is controlled by post-transcriptional gene regulation. Specifically, components of the uPA system, NKG2D ligands, and the cytokines IFN-γ, GM-CSF, IL-10, TNF-α, and MIP-1α are post-transcriptionally regulated, mainly by mRNA stabilization/destabilization or translational control. These control mechanisms are not only essential for maintaining proper protein levels, but also allow for fast and sensitive responses of NK cells to target cells.
Although the role of noncoding RNAs in immune responses is an emerging hot topic, the regulation of NK cell functions is poorly understood. Moreover, few studies on post-transcriptional regulation during NK cell development have been reported. A better understanding of gene regulation at the post-transcriptional level during NK cell differentiation and activation may assist in the development of novel NK cell-based immunotherapies for major human diseases.

 XML Download
XML Download