

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Mosquito-borne flaviviruses, including Japanese encephalitis (JE), West Nile (WN), and Zika viruses, are major current and emerging threats to millions of people worldwide. Among the neurotropic flaviviruses, JE virus (JEV) is considered the most serious because of its neurotropical impacts, higher fatality rate, and permanent neuropsychiatric sequelae (1). The expanding geographical range of JEV and potential for outbreaks of JE in nonimmune populations demand an urgent effort to develop effective antiviral therapies or alternate strategies to limit the immunopathology of JE (2). Indeed, considerable progress in immunopathological studies of JE has been made in both infected patients and murine models (345678). JEV replicates in monocytes/macrophages and dendritic cells (DCs) as primary target cells (9), and in these cells, the virus travels from the periphery to the central nervous system (CNS), where it causes neurological disorders (10).
DCs are antigen-presenting cells (APC) that play a crucial role in initiating adaptive immune responses as well as regulating innate immunity, with various immunological functions such as the ability to express MHC class II, stimulate naïve T-cells, and crosstalk with NK cells (11121314). The role of DCs in neuroinflammation caused by neurotropic viruses like JEV is not fully understood because of their complex multi-cellular interactions with innate-like NK cells, monocytes, and macrophages and functions in immune homeostasis and tolerization (1516). However, CD11c-DTR transgenic (Tg) mice, which express the diphtheria toxin receptor (DTR) gene under the control of a cloned Itgax promoter and allow conditional DC depletion upon DT injection, have been a critical tool in the in vivo study of DC immunology (17). Using this conditional DC depletion model, we previously demonstrated marked exacerbation of JE progression and abnormal differentiation of inflammatory CD11b+Ly-6Chi monocytes following CD11chi DC ablation (18). Moreover, CD11chi DC ablation enhanced the permeability of the blood-brain barrier (BBB) by affecting the regulation of tight junction and adhesion molecules during JE progression (19).
Typically, murine monocytes are subdivided by the expression of Ly-6C and CX3CR1 into Ly-6ChiCX3CR1lo CCR2+CD62L− and Ly-6CloCX3CR1hiCCR2−CD62L−monocytes. CD11b+Ly-6Clo monocytes are known to play a role in regulating host defense and repairing tissues after inflammatory injury, whereas CD11b+Ly-6Chi monocytes are specifically recruited by CCL2 to inflamed sites, where they become classically activated M1 macrophages and/or Tip-DCs (20). Thus, CD11b+Ly-6Chi monocytes are known to significantly reduce the immune-privileged status of the CNS (21) and exacerbate the pathogenesis of viral encephalitis (182223). In addition, CD4+Foxp3+ regulatory T cells (Tregs), which regulate excessive immune responses, accumulate preferentially over other effector Th cells at inflamed sites because of homing receptors such as CCR5 (242526). In contrast, a skewed IL-17+CD4+ Th17 cell response at inflamed sites may exacerbate JE progression (26). Results of these studies provide insight into how the balance of pro- and anti-inflammatory leukocytes can affect the progression of immunopathological diseases such as JE.
Here, we examined the changes in lymphoid and myeloidderived leukocyte subpopulations related to pro- and anti-inflammatory reactions during JE progression in CD11chi DC-ablated mice, with a focus on the infiltration of Foxp3+ Treg/IL-17+ Th17 cells and Ly-6Chi/Ly-6Clo monocytes into lymphoid tissue and the CNS. Our data revealed that CD11chi DC ablation resulted in the predominance of IL-17+CD4+ Th17 cells and inflammatory CD11b+Ly-6Chi monocytes over Foxp3+ Tregs and CD11b+Ly-6Clo monocytes in the lymphoid tissue and CNS during JE progression. Therefore, the imbalanced environment of Foxp3+ Treg/IL-17+ Th17 cells and Ly-6Chi/Ly-6Clo monocytes in response to CD11chi DC ablation may contribute to the exacerbation of JE progression.
MATERIALS AND METHODS
Ethics statement
All animal experiments described in the present study were conducted at Chonbuk National University according to the guidelines set by the Institutional Animal Care and Use Committees (IACUC) of Chonbuk National University (http://lac.honamlife.com/research/research_05.php). The study was pre-approved by the Ethical Committee for Animal Experiments of Chonbuk National University (permission code 2013-0028). Animal research protocols used in this study followed the guideline set up by the nationally recognized Korea Association for Laboratory Animal Sciences (KALAS). All experimental protocols requiring biosafety were approved by the Institutional Biosafety Committees (IBC) of Chonbuk National University.
Animals, cells, viruses, and reagents
C57BL/6 (H-2b) mice (4~6 weeks old) were purchased from Samtako (O-San, Korea). CD11c-DTR transgenic (Tg) mice (B6.FVB-Tg Itgax-DTR/EGFP 57Lan/J [DTR]), which express the diphtheria toxin (DTR) gene under control of a cloned Itgax promoter and thus allow conditional DC depletion upon DT injection (17), were obtained from Jackson Laboratories (Bar Harbor, ME, USA). All mice were genotyped and bred in the animal facilities of Chonbuk National University. The JEV Beijing-1 strain was propagated in a mosquito cell line (C6/36), using DMEM supplemented with 2% FBS, penicillin (100 U/ml), and streptomycin (100 U/ml) (1819). The virus stocks were titrated by conventional plaque assay using BHK-21 cells (CCL-10; American Type Culture Collection), and stored in aliquots at -80℃ until use. The mAbs used for the flow cytometric analysis and other experiments were obtained from eBioscience (San Diego, CA, USA) or BD Biosciences (San Diego, CA, USA). Diphtheria toxin (DT) was purchased from Sigma-Aldrich (St. Louis, MO, USA). The primers specific for cytokines (181926) were synthesized at Bioneer Corp. (Daejeon, Korea) and were used for PCR amplification of target genes.
Intracellular staining for analysis of CD4+ Th1, Th17, and Treg cells
To monitor CD4+ Th cell subsets, mice were infected i.p. with 3.0×107 pfu of JEV and sacrificed at 5 days postinfection (dpi). Brain leukocytes and splenocytes were prepared and cultured in 96-well plates (106 cells/well) with PMA plus ionomycin (Th1 and Th17) in the presence of monensin (2 µM) at 37℃ for 5 h. Stimulated cells were washed twice with PBS and surface-stained with FITC-anti-CD4 at 4℃ for 30 min. After washing twice with PBS containing monensin and then fixing cells, cells were washed twice with permeabilization buffer (eBioscience, San Diego, CA) and then stained with PerCP-anti-IFN-γ and APC-anti-IL-17α in permeabilization buffer at room temperature for 30 min. After washing twice with PBS, cells were fixed with fixation buffer. To monitor Tregs, brain leukocytes and splenocytes were surfacestained with FITC-anti-CD4 markers on ice for 30 min, followed by fixation with permeabilization concentrate buffer (eBioscience, San Diego, CA) at 4℃ for 6 h. After fixation, cells were washed twice with permeabilization buffer and stained with PE-anti-Foxp3 antibody in permeabilization buffer at room temperature for 30 min. Samples were analyzed with a FACSCalibur flow cytometer. To monitor the expression of transcription factors specific for each Th cell subset, CD4+ T cells were sorted from CNS-infiltrated leukocytes and briefly stimulated with PMA plus ionomycin for 3 h. The expression of transcription factors (RORγT, T-bet, GATA3, Foxp3) specific for each Th cell subset was determined by real-time quantitative RT-PCR (qRT-PCR), using total RNA extracted from stimulated CD4+ T cells.
Analysis of splenic and CNS-infiltrated monocytes
Mice infected with JEV were perfused with 30 ml of HBSS at 3 dpi via cardiac puncture of the left ventricle. Brains were harvested and homogenized by gently pressing them through 100-mesh tissue sieves and then digested with 25 µg/ml collagenase type IV (Worthington Biochem, Freehold, NJ), 0.1 µg/ml trypsin inhibitor Nα-p-tosyl-L-lysine chloromethyl ketone, 10 µg/ml DNase I (Amresco, Solon, OH), and 10 mM HEPES in HBSS at 37℃ for 1 h with shaking. Cells were separated using Optiprep density gradient (18/10/5%) centrifugation at 800×g for 30 min (Axis-Shield, Oslo, Norway), and then cells were collected from the 18% to 10% interface and washed twice with PBS. Splenocytes and brain leukocytes were then counted and stained for CD11b, Ly6G, Ly6C, and CD45, using directly conjugated antibodies (eBioscience), at 4℃ for 30 min. Finally, these cells were fixed with 10% formaldehyde. Data were collected and analyzed with a FACSCalibur flow cytometer (Becton Dickson Medical Systems, Sharon, MA) and FlowJo (Tree Star, San Carlos, CA) software, respectively.
Real-time qRT-PCR
The expression of cytokines (IL-1β, TNF-αIL-6, IL-10, TGF-β) and growth factors (Flt3-L, G-CSF) in sorted CD11b+Ly-6Clo monocytes was determined by quantitative SYBR Green-based real-time qRT-PCR. Mice were infected i.p. with JEV (3.0×107 PFU), and splenic CD11b+Ly-6Clo monocytes were sorted at 3 dpi. Total RNAs were extracted from sorted CD11b+Ly-6Clo monocytes and then used in real-time qRT-PCR performed with a CFX96 Real-Time PCR Detection system (Bio-Rad Laboratories, Hercules, CA, USA). Following reverse transcription of total RNAs using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA), a reaction mixture containing 2 µl of template cDNA, 10 µl of 2× SYBR Primix Ex Taq, and 500 nM primers in a final volume of 20 µl was prepared. The cDNA was denatured at 95℃ for 30 s and then subjected to 45 cycles of 95℃ for 5 s and 60℃ for 20 s. After the reaction was completed, the temperature was increased from 65℃ to 95℃ at a rate of 0.2℃/15 s, and fluorescence was measured every 5 s to construct a melting curve. A control sample that contained no template DNA was run with each assay, and all reactions were performed at least in duplicate to ensure reproducibility. Amplification of the target was confirmed by a melting curve analysis. All data were analyzed using Bio-Rad CFX Manager version 2.1 software (Bio-Rad Laboratories).
Statistical analysis
All data were expressed as the average±standard error, and statistically significant differences between groups were analyzed by unpaired two-tailed Student's t-test for leukocyte population analysis or ANOVA and post-hoc testing for multiple comparisons of the mean. The statistical significance of cytokine gene expression was evaluated by Mann-Whitney test or unpaired two-tailed Student's t-test. A p-value≤0.05 was considered significant. All data were analyzed using Prism software (GraphPadPrism4, San Diego, CA, USA).
RESULTS and discussion
Increase of IL-17+CD4+ Th17 cell proportion following CD11chi DC ablation in lymphoid tissues and the CNS during JE progression
We previously showed that CD11chi DC ablation exacerbated JE progression and resulted in the under-maturation of CD11b+Ly-6Chi monocytes and enhanced permeability of the BBB (1819). Here, we hypothesized that JE was exacerbated in CD11chi DC-ablated mice through additional mechanisms such as abnormal infiltration and/or maturation of lymphoid and myeloid leukocytes related to pro- and anti-inflammatory processes. Since the roles and quantities of CNS-infiltrated CD4+ Th cell subsets, including IFN-γ+ Th1, IL-17+ Th17, and Foxp3+ Tregs, have been not defined, we decided to examine changes in CD4+ Th cell subsets in the spleens and brains of CD11chi DC-ablated mice during JE progression. Interestingly, our data revealed that CD11chi DC-ablated mice showed a higher frequency of IL-17+CD4+ Th17 cells in spleens at 5 dpi, but IFN-γ+CD4+ Th1 cells showed no change in frequency (Fig. 1A). The accumulation of IL-17+CD4+ Th17 cells in spleens was more apparent in CD11chi DC-ablated mice; CD11chi DC-ablated mice contained fewer IFN-γ+CD4+ Th1 cells in their spleens (Fig. 1B). In addition, CD4+Foxp3+ Treg cells were detected at a lower frequency in CD11chi DC-ablated mice during JE progression (Fig. 1C), and CD11chi DC-ablated mice had fewer CD4+Foxp3+ Tregs in their spleens (Fig. 1D). These data suggest that the altered balance of CD4+ Th cell subsets (IL-17+ Th17 and IFN-γ+ Th1 cells) and regulatory CD4+Foxp3+ Tregs may contribute to the exacerbation of JE in CD11chi DC-ablated mice. Furthermore, the frequency and total number of infiltrated IL-17+ Th17, IFN-γ+ Th1, and Foxp3+ Treg cells were determined in the CNS at 5 dpi. As expected, IL-17+CD4+ Th17 cells were detected at higher levels in CD11chi DC-ablated mice (Fig. 1E and F). However, CD11chi DC-ablated mice showed a higher frequency and absolute number of IFN-γ+ CD4+ Th1 cells in the CNS, which contrasted with the results from the spleen. This discrepancy may be caused by CNS-infiltration of CD4+ Th1 cells from the spleen that is considered reservoir. Therefore, enhanced CNS-infiltration of Th17 and Th1 cells seems to contribute to the exacerbation of JE in CD11chi DC-ablated mice. In support of this, vehicle-treated CD11c-DTR mice showed higher proportion of CNS-infiltrated CD4+Foxp3+ Treg cells (both frequency and total number) than CD11chi DC-ablated mice (Fig. 1G and H). This imbalance in Th17 and regulatory Treg cells that infiltrated the CNS resulted in a higher ratio of Tregs to Th17 cells in vehicle-treated CD11c-DTR mice, compared to that in CD11chi DC-ablated mice (Fig. 1I).
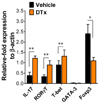
Moreover, we examined the expression of transcription factors associated with the differentiation of CD4+ Th cell subsets in CD4+ T cells sorted from the brain. As expected, CD4+ T cells from the brains of CD11chi DC-ablated mice showed higher levels of IL-17 and RORγT in the CD4+ Th17 cell subset and T-bet in the CD4+ Th1 cell subset, whereas Foxp3 expression in Tregs was lower (Fig. 2). In conclusion, CD11chi DC ablation causes an imbalance in Th17, Th1, and Treg CD4+ cell subsets during JE progression, thereby contributing to the exacerbation of JE.
CD11chi DC ablation causes fewer Ly-6Clo monocytes to accumulate in lymphoid tissues and the CNS during JE progression
Myeloid-derived leukocytes in the CNS may be responsible for inducing pro- and anti-inflammatory changes, which can lead to significant pathological outcomes during JE progression. Monocytes and macrophages are important contributors and/or regulators of immunopathological progression. Typically, two principle subsets of murine monocytes are identified, based on the expression of lymphocyte antigen 6C (Ly-6C) (20). Inflammatory/classical monocytes are Ly-6ChiCCR2hiCX3CR1lo/int, whereas patrolling/non-classical monocytes are Ly-6Clo/intCCR2–CX3CR1hi (20). In a previous study, we showed that CD11chi DC ablation caused abnormal differentiation of inflammatory CD11b+Ly-6Chi monocytes during JE progression, contributing to the exacerbation of JE (18). Therefore, we were interested in the changes in CD11b+Ly-6Clo monocytes, but not CD11b+Ly-6Chi monocytes, during JE progression in CD11chi DC-ablated mice. CD11chi DC ablation increased the proportion of inflammatory CD11b+Ly-6Chi monocytes in spleens during JE progression, whereas CD11b+Ly-6Clo monocytes decreased in CD11chi DC-ablated mice, compared to those in vehicle-treated CD11c-DTR mice (Fig. 3A). Accordingly, CD11chi DC-ablated mice contained a higher number of inflammatory CD11b+Ly-6Chi monocytes in the spleen, but a lower number of splenic CD11b+Ly-6Clo monocytes (Fig. 3B). In line with the data from the spleen, CD11chi DC-ablated mice showed a higher frequency and total number of CNS-infiltrated CD11b+Ly-6Chi monocytes, but fewer CD11b+Ly-6Clo monocytes in the brain (Fig. 3C and D). This contrast in the accumulation of CD11b+Ly-6Chi and Ly-6Clo monocytes in the spleens and brains of CD11chi DC-ablated mice resulted in a markedly increased ratio of Ly-6Chi to Ly-6Clo monocytes (Fig. 3E). This dysregulated accumulation of CD11b+Ly-6Chi and Ly-6Clo monocytes in the spleen and brain may contribute to the exacerbated progression of JE in CD11chi DC-ablated mice.
Altered features of CD11b+Ly-6Clo monocytes in CD11chi DC-ablated mice
CD11chi DC ablation induces the abnormal differentiation of inflammatory CD11b+Ly-6Chi monocytes and thereby exacerbates JE progression (18). Thus, we examined the differentiation of CD11b+Ly-6Clo monocytes in CD11chi DC-ablated mice during JE progression. Our data revealed that CD11b+Ly-6Clo monocytes from the spleens of CD11chi DC-ablated mice expressed anti-inflammatory cytokines such as IL-10 and TGF-β at lower levels, compared to those from CD11b+Ly-6Clo monocytes in vehicle-treated CD11c-DTR mice (Fig. 4A). One interesting result was that splenic CD11b+Ly-6Clo monocytes from vehicle-treated CD11c-DTR mice showed higher levels of expression of some growth factors associated with the differentiation of monocytes and DCs, including G-CSF and Flt3-L. Furthermore, CD11b+Ly-6Clo monocytes in the spleens of CD11chi DC-ablated mice showed lower levels of expression of activation markers, compared to those in vehicle-treated CD11c-DTR mice (Fig. 4B). These data indicate that CD11chi DC ablation can cause under-differentiation of CD11b+Ly-6Clo monocytes during JE progression.
In conclusion, our data demonstrate that CD11chi DC ablation results in the imbalanced accumulation of CD4+ Th17/Treg cells and CD11b+Ly-6Chi/Ly-6Clo monocytes in the lymphoid tissue and CNS during JE progression. It is unclear whether this imbalanced infiltration of Th17/Treg cells and Ly-6Chi/Ly-6Clo monocytes was directly caused by CD11chi DC ablation, or whether CD11chi DC ablation induced an imbalance in Th17/Treg cells and Ly-6Chi/Ly-6Clo monocytes through secondary pathologic pathways in exacerbated JE progression. However, theoretically, it is conceivable that enhanced infiltration of IL-17+CD4+ Th17 and inflammatory CD11b+Ly-6Chi monocytes in the CNS facilitates JE progression (1826). Our data supports this, suggesting that CD11chi DC ablation can exacerbate JE progression by promoting an imbalance in the infiltration of IL-17+Th17 to Foxp3+ Treg cells and Ly-6Chi to Ly-6Clo monocytes in the CNS. Furthermore, CD11b+Ly-6Chi monocytes likely differentiate into CD11b+Ly-6Clo monocytes and become tissue-resident macrophages at the late stage in inflammatory disease (15). Thus, the ablation of CD11chi DCs, which share precursors with cells in the monocyte-macrophage lineage, might affect the differentiation of CD11b+Ly-6Chi and Ly-6Clo monocytes in inflamed tissues. Our previous study showed that CD11chi DC ablation resulted in under-differentiated phenotypes of inflammatory CD11b+Ly-6Chi monocytes during JE progression (18). Similarly, the present results indicate additional regulation of CD11b+Ly-6Clo monocyte differentiation in response to CD11chi DC ablation, resulting in decreased expression of anti-inflammatory cytokines (IL-10 and TGF-β) that limits excess inflammation. This inappropriate differentiation of CD11b+Ly-6Clo monocytes in CD11chi DC-ablated mice may fail to regulate JE progression. Collectively, data in this study suggest that CD11chi DC ablation induced an imbalance in IL-17+ Th17 and Foxp3+ Treg cells and abnormally differentiated CD11b+Ly-6Chi to Ly-6Clo monocytes during JE progression, thereby facilitating JE progression.



 XML Download
XML Download