

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cortisol is a glucocorticoid hormone (GH) mainly produced in the adrenal gland and is tightly regulated via the hypothalamic-pituitary-adrenal (HPA) axis (1). Besides, it can be converted from its substrate cortisone by the action of 11β-hydroxysteroid dehydrogenase (11β-HSD1) in the peripheral tissues, including the liver, fat, brain, and skeletal muscle (2345). Physiologically, cortisol is a biologically active GH found in humans, and exerts pleiotropic effects by binding to the glucocorticoid receptor (GR) (67). Blood cortisol levels are closely associated with the development of diseases, such as Cushing's syndrome and Addison's disease (89). It has also been reported that dysregulation of cortisol is closely linked to metabolic syndromes, including type 2 diabetes and obesity. On the contrary, cortisol exhibits a potent anti-inflammatory activity within the normal range, although the precise mechanisms remain unclear. In fact, cortisol acts as a regulator of lymphocytes, which are recruited to the injury sites to trigger inflammation (10). It can be mechanistically proposed that immunosuppressive or anti-inflammatory effects of cortisol are accomplished by blocking inflammatory signals from the transcription factors, especially nuclear factor-κB (NFκB) and AP-1 (11).
Rheumatoid arthritis (RA) is characterized by several changes, including synovial hyperplasia, inflamed synovial membrane, leukocyte recruitment, and cytokine overproduction, resulting in the formation of pannus—abnormal layers of granulation tissue in which massive inflammatory signals occur. Synovial membrane consists of macrophage-like synovial cells and fibroblast-like synoviocytes (FLS), which are involved in maintenance of the internal joint homeostasis. In particular, FLS play key roles in the pathogenesis of RA by producing various chemokines and matrix metalloproteinases (MMPs) (12). A human synovial cell line, SW982, was used for investigating the anti-inflammatory effects of GH. SW982 cells were previously reported to express 11β-HSD1, but not 11β-HSD2 (13), that can catalyze its substrate cortisone into cortisol. Accordingly, this study aimed to investigate the regulation of the inflammatory responses of synovial cells to external stimuli by autocrine glucocorticoids (GCs).
Primarily, we optimized various conditions, such as substrate concentration, cell seeding density, and incubation period that would affect the conversion of inactive substrate to active product in SW982 cells. SW982 cells can be grown in monolayer or spheroid conditions that simulate inflamed synovium. However, there was a slight difference between the 11β-HSD1 activity of the monolayer culture and that of the spheroid culture. To examine the effects of 11β-HSD1 on inflammatory responses, monolayer culture of SW982 was stimulated with lipopolysaccharide (LPS) with or without cortisone. Cortisol produced by cellular 11β-HSD1 downregulated LPS-mediated cytokines, including interleukin (IL)-β and IL-6. Treatment of cells with 11β-HSD1 inhibitor or GH antagonist reversed anti-inflammatory effects of cortisol, suggesting that its mode of action is closely correlated to prereceptor regulation. Furthermore, we suggested that cortisol could inactivate LPS-mediated NFκB activation via regulation of IκB phosphorylation. In summary, locally regenerated cortisol level might alleviate various inflammatory genes under NFκB regulation, highlighting that prereceptor regulation of 11β-HSD1 is significant in the pathogenesis of inflammation-associated diseases.
MATERIALS AND METHODS
Materials
Cortisone, cortisol, carbenoxolone (CBX), and 18β-glycyrrhetinic acid (GA) were obtained from Sigma (St Louis, MO). Enhancer spray, and [3H] cortisone and cortisol as radioactive substrate and product were obtained from Amersham Life Sciences Ltd. (Piscataway, NJ). HTRF® cortisol assay product was obtained from Nihon Schering K.K. (Tokyo, Japan). EcoDye DNA staining solution was purchased from Biofact (Daejeon, South Korea). IκB and phosphor-IκB were purchased from Cell Signaling Technology (Danvers, MA). All other chemicals and reagents were purchased from Sigma-Aldrich.
Cell culture conditions
Cell line SW982 (ATCC:HTB-93) was purchased from ATCC and maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with antibiotics and 10% fetal bovine serum (FBS). Conventionally, cells were seeded into tissue culture plates and incubated until attachment to plate base. To simulate pannus condition, cells were plated at a high density into each well of 96-well plate, the bottoms of which were not pretreated, and incubated for at least 4 days.
Measurement of cellular 11β-HSD catalytic activity
To verify the catalytic conversion from cortisone to cortisol, enzyme activity was also measured by conversion of radioactive cortisone into cortisol on TLC [8]. [3H] Cortisone (160 nM, specific activity: 42.0 Ci/mmol) was added to SW982 culture (500 µL) grown in 24-well plates and incubated for given times. Cortisone metabolites present in culture medium were extracted with ethyl acetate and separated by thin layer chromatography (TLC) using chloroform:methanol (9:1, v/v) as development solution and then visualized by autoradiography of the plate sprayed with enhancer on film. In contrast, non-radioactive assay was carried out by HTRF® protocol. Briefly, cells were seeded onto 24-well plates at different densities and were incubated in 500 µL DMEM containing 160 nM cortisone. Culture media (2 µL) were removed at the indicated time points, and enzyme activity was assessed by HTRF® assay as carried out previously (14).
Detection of inflammatory cytokines induced by LPS stimulation
SW982 cells were stimulated with LPS and then the level of its downstream inflammatory cytokines were monitored and correlated with the levels of 11β-HSD1 expression. RNAs extracted from SW982 cells were amplified by reverse transcription-polymerase chain reaction (RT-PCR) using specific primers described below. Specific primers for IL-1β, IL-6, 11β-HSD1, and β-actin were designed for amplification of these genes. Sequences for those genes are as follow. S or AS in parentheses indicates sense or antisense primer, respectively.
IL-6: 5′-ATG AAC TCC TTC TCC ACA AGC GC (S)-3′, 5′-GAA GAG CCC TCA GGC TGG ACT G-3′ (AS)
IL-β: 5′-AAA CAG ATG AAG TGC TCC TTC CAG G-3′(S), 5′-TGG AGA ACA CCA CTT GTT GCT CCA-3′ (AS)
11β-HSD1: 5′-TTG CTT TGG ATG GGT TCT TC-3′ (S), 5′-AGA GCT CCC CCT TTG ATG AT-3′ (AS)
β-actin: 5′-GCC ATG TAC GTT GCT ATC-3′ (S), 5′-CTC CTT AAT GTC ACG CAC-3′(AS)
Aliquots of the PCR products were electrophoresed on 1.2% agarose gel, followed by visualization using EcoDye staining solution, and gel images were acquired with a UV trans-illuminator. To investigate the effects of LPS alone or LPS with cortisone on inflammatory cytokines, RNA samples were extracted from cells treated with LPS in the presence or absence of cortisone and subjected to RT-PCR using specific primers for IL-6 and IL-1β.
Changes in NFκB activity of SW982 cells treated with LPS in the presence of cortisone and cortisol
NFκB activity was indirectly determined by measuring the levels of phosphor-IκB. The lysates prepared from SW982 cells were subjected to SDS-PAGE and transferred onto nitrocellulose (NC) membrane. Resolved proteins on NC were probed with an antibody against phosphor-IκB, and then further incubated with a secondary antibody conjugated with horseradish peroxidase to visualize its target proteins.
RESULTS
Optimization of conditions for catalytic activity of 11β-HSD1 in SW982 cells
The catalytic activity of cellular 11β-HSD1 in SW982 cells to convert cortisone into cortisol was examined on TLC. Incubation of SW982 cells with cortisone as a substrate could convert cortisone into cortisol in a cell density dependent manner as demonstrated by slower migration of products than reactants on a TLC plate (Fig. 1A). HTRF assay, a non-radioactive method, was employed to detect cellular 11β-HSD1 activity. As shown in Fig. 1B, conversion of cortisone into cortisol was proportional to cell numbers plated, indicating that cells at a density of 106 converted most of the cortisone within 48 h. Moreover, cortisone was converted into cortisol in cells in a time dependent manner. To ascertain the involvement of cellular 11β-HSD1 in cortisone/cortisol conversion, 11β-HSD inhibitors were used. Treatment of cells with either GA or CBX substantially inhibited cortisol production, suggesting that cellular 11β-HSD1 is responsible for catalytic synthesis of cortisol (Fig. 1C).
11β-HS D1 activity in SW 982 cells under monolayer and spheroid cultures
SW982 cells can be grown in two different conditions—monolayer and spheroid cultures (Fig. 2A). Microscopic observation revealed that spheroid culture induced conglomeration of cells within a limited area. 11β-HSD1 activity of cells grown under these culture conditions was compared using the HTRF assay, and a slight difference in the cortisol production was noted between monolayer and spheroid cultures (Fig. 2B).
Effects of various stimulants on 11β-HSD1 activity
The catalytic activity of 11β-HSD1 was monitored after SW982 cells were stimulated with pro-inflammatory cytokines or LPS (Fig. 3). Cells treated with IL-1β, TNFα, or LPS (concentrations lower than 100 ng/mL) did not substantially affect 11β-HSD1 activity, while those treated with LPS at a concentration of 100 ng/mL significantly reduced its activity by 30%.
Effect of cellular 11β-HSD1 activity on LPS-stimulated inflammatory responses
As mentioned previously, expression levels of cytokines after treatment of cells with LPS were preliminarily evaluated by RT-PCR (Fig. 4). Stimulation of cells with LPS induced pro-inflammatory cytokines, IL-1β and IL-6. Addition of cortisone into SW982 culture suppressed LPS-induced inflammatory responses. Conversely, either GA or RU486 reversed the anti-inflammatory effects of cortisone against LPS stimulation.
Suppression of NFκB activation by active cortisol or cortisone pre-incubation in LPS-stimulated SW982 cells
It has been known that LPS induces various inflammatory cytokines via NFκB activation, and that cortisol has a negative effect on LPS-stimulated inflammation via its cognate receptor. To better understand the mechanism underlying suppression of various inflammatory cytokines by active cortisol, NFκB activity was investigated following LPS stimulation in the absence or presence of cortisone (Fig. 5). NFκB activation was indirectly evaluated by phosphor-IκB levels in SW982 cells. Cells showed NFκB activation in response to LPS, while LPS-induced NFκB activation was suppressed in a dose-dependent manner.
DISCUSSION
Synovial cells, SW982, express 11β-HSD type 1, but not type 2, that catalyzes the conversion of cortisone to cortisol. The principal role of cortisol is to increase glucose level by gluconeogenesis and carbohydrate metabolism (1516). Additionally, cortisol can be engaged in the suppression of acute immune responses and in the reduction of bone formation (17). Specifically, numerous lines of evidence suggest that inflammatory activation of synovial cells has been decisive in the pathogenesis of degenerative arthritis. Therefore, this study reveals the mechanism underlying regulation of the LPS-induced inflammatory responses by 11β-HSD activity. Autocrine cortisol significantly suppressed various LPS-stimulated responses, indicating that 11β-HSD might effectively regulate the local inflammatory responses.
Since GCs activate the negative feedback circuit of inflammation, they are widely prescribed in medicine to treat various diseases, such as allergies, asthma, autoimmune diseases, and sepsis (18192021). Furthermore, GC at high doses can reject the abnormal cancer cells helping towards treating cancers. However, their overuse should be restricted because of diverse pleiotropic effects on metabolic homeostasis as well as the immune system. Mechanistically, GCs exhibit their effects by binding to the GR, not only by trans-activating anti-inflammatory proteins, but also by trans-repressing pro-inflammatory proteins, to downregulate inflammation (22). In fact, an active GC cortisol inhibits T-cell proliferation by desensitizing T-cells to IL-1 and suppressing synthesis of T-cell growth factor (23). Moreover, cortisol synthesis in the epidermis exhibits a negative feedback effect on inflammatory cytokine IL-1 upon tissue injury (24).
Previously, it was reported that SW982 cells were phenotypically characterized after stimulation with IL-1β in the presence of dexamethasone (25). Dexamethasone effectively downregulates IL-1β, IL-6, and COX-2 transcripts at high cell density, but not at low cell density. To examine the anti-inflammatory role of locally regenerated cortisol, in this study, SW982 cells were stimulated with LPS in the presence or absence of cortisone. The conversion reaction between cortisone and cortisol was catalyzed in the forward and reverse directions by 11β-HSD1 and 11β-HSD2, respectively, but the latter was a more potent GC. It has been already reported that SW982 cells express 11β-HSD1, but not 11β-HSD2. 11β-HSD1 in SW982 cells catalyzes conversion of cortisone into cortisol, an active GH. Its cellular conversion was verified from TLC analysis of radioactive products present in culture media of SW982 cells (Fig. 1). The use of radioactive cortisone for TLC analysis was time-consuming and labor-intensive; therefore, a non-radioactive method was adopted to monitor the levels of cortisol, without its purification. An increase in cortisol production corresponding to the cell seeding density was comparable to values obtained from the HTRF assay, which depends on the use of an antibody specific towards cortisol. Cells were further optimized for incubation period and substrate concentration required to convert cortisone into cortisol during culture. The catalytic requirement of 11β-HSD1 for metabolic conversion was appropriately proved by the fact that cortisol synthesis was inhibited by an inhibitor of 11b-HSD1, CBX or GA. Notably, SW982 cells were grown in two different culture conditions—monolayer and spheroid cultures. Unlike monolayer culture of SW982 cells, spheroid culture mimics active FLS in the pannus of patients with RA. Since there was no significant difference in 11β-HSD1 activity between both the culture conditions (Fig. 3), SW982 cells grown under monolayer condition were treated with cytokines or LPS to provoke an inflammatory response. IL-1β or TNFα did not significantly increase 11β-HSD1 expression, while LPS at a high dose decreased 11β-HSD1 transcript in SW982 cells (Fig. 3). Pro-inflammatory cytokines, such as IL-1α/TNFα, increase 11β-HSD1 in human epithelial cells, A549, via phosphorylation of C/EBPβ (26) and keratinocytes (27). Conversely, it has been reported that 11β-HSD1-catalyzed cortisol production modulates expression of inflammatory cytokines.
LPS induced various inflammatory outcomes similar to those in previous reports, while pretreatment of SW982 cells with cortisone dramatically abrogated such LPS-induced inflammatory responses (Fig. 4). Cortisol is known as a potent anti-inflammatory hormone, and its underlying mechanisms have been proposed. Cortisol treatment suppresses LPS-induced IL-6, but not IL-8, transcripts (28). Generally, its anti-inflammatory potency is attributed to negative transcriptional regulation of the inflammatory gene (29). In fact, cortisol binds to the cognate GR in the cytoplasm to neutralize NFκB, which induces gene expression of inflammation-related proteins and adherence factors (30). Additionally, it partially interferes with a heterodimeric protein, AP-1, composed of Fos and Jun, which can induce transcriptional activation of pro-inflammatory proteins (31). Consequently, cortisol synthesized from cortisone was proposed to reduce LPS-mediated inflammatory responses through NFκB or AP-1 transcriptional regulation.
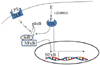
In summary, cortisol production catalyzed by a cellular enzyme prevents LPS-derived inflammatory response in the cell (Fig. 6). LPS induces pro-inflammatory cytokines by binding to TLR4 and then activating NFκB. Production of autocrine cortisol from cortisone is driven by cellular 11β-HSD1 and resulting locally regenerated cortisol might interfere with NFκB activation directly or indirectly, synergistically suppressing inflammatory responses to LPS. In conclusion, these results highlight that levels of locally regenerated cortisol in synovial cells play a key role in modulating inflammatory response to LPS or cytokines in synovial cells, which could cause pathogenesis of RA.





 XML Download
XML Download