

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Allogeneic (allo) hematopoietic stem cell transplantation (HSCT) is an effective treatment for hematological disorders, including lymphoma and leukemia (1234). Graft-versus-leukemia (GVL) effects, which are derived from the activation of donor T cells that recognize the allo-antigens expressed by the recipient's tumor cells, contribute to the eradication of malignant host cells (5). However, donor T cells are also reactive to allo-antigens expressed by the recipient's tissues and parenchymal cells in the gastrointestinal (GI) tract, liver, lung, and skin, and induce graft-versus-host disease (GVHD), a life-threatening complication of allo-HSCT (67). The suppression of severe GVHD is important for the success of allo-HSCT.
GI tract damage is a critical event in the pathogenesis of GVHD (89). The integrity of the GI tract and innate immunity to the intestinal microbiome both contributes to the maintenance of intestinal homeostasis; disruption of intestinal homeostasis during allo-HSCT provokes intestinal GVHD, which leads to exacerbation of the disease and systemic GVHD (9). Signaling through Toll-like receptors (TLRs) and myeloid differentiation factor 88 (MyD88), a signaling adaptor downstream of TLRs, is pivotal in innate immunity that controls response to microbial stimulation; evidence supporting the significances of their signaling in GVHD is accumulating (1011). In this article, we will review recent research into the role of TLR/MyD88-mediated innate immunity in acute intestinal GVHD.
ACUTE AND CHRONIC GVHD
GVHD is broadly classified into acute and chronic GVHD, depending on the timing of disease incidence after allo-HSCT. Chronic GVHD was classically defined as a late complication of allo-BMT that occurs in 100 days post-transplantation. Chronic GVHD is similar to autoimmune and other immunological diseases, such as scleroderma (1213), systemic lupus-like diseases (14), primary biliary cirrhosis (15), and immune cytopenia (16); it is characterized by tissue inflammation and fibrosis, and is mediated by cellular and CD4 T helper cell type 2-dependent humoral immunity (1718). In 2014, revised chronic GVHD criteria were proposed, which facilitate distinction of chronic and acute GVHD, that include diagnostics in the skin (e.g., poikiloderma and sclerotic features including lichen planus-like features), mouth (e.g., lichen planus-like changes), lung (e.g., bronchiolitis obliterans), and GI tract (e.g., esophageal web, strictures or stenosis in the upper to middle third of the esophagus) (19).
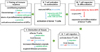
Development of acute GVHD is observed within 100 days post-HSCT, with symptoms indicating damage to the skin (e.g., maculopapular rash on the palms, soles and ears, and diffuse erythematous rash over the entire body), liver (e.g., hyperbilirubinemia, jaundice, and elevated transaminases), GI tract (e.g., nausea, vomiting, abdominal cramps, anorexia, bleeding, and diarrhea), and, occasionally, lungs, eyes and oral mucosa (20). Although donor T cell-mediated adaptive immunity is an essential component of the development of acute GVHD, innate immunity also plays significant roles (62122). Chemoirradiation conditioning of recipients prior to HSCT provokes apoptosis of epithelial cells and tissue inflammation in several organs, including the intestines. The release of inflammatory cytokines activates antigen-presenting cells (APCs), which promote the activation and effector differentiation of allo-reactive donor T cells. Activated of T cells mediate cytotoxicity against allo-antigen-bearing recipient cells in damaged tissues, which increase inflammation in the target organs (Fig. 1). In particular, intestinal inflammation initiated by epithelial cell damage disrupts the epithelial barrier, which exposes innate immune cells to intestinal microbial stimuli. This innate cell stimulation by microbial antigens enhances the recruitment of activated T cells to the intestines, where they kill GI epithelial cells and cause cryptic cell degeneration, resulting in heightened intestinal inflammation and nutrient malabsorption. The degree of intestinal inflammation is associated with the severity of acute GVHD. Acute intestinal GVHD occurs in more than 50% of allo-HSCT patients (23).
GUT MICROBIOME AND INNATE IMMUNITY IN ACUTE INTESTINAL GVHD
The gut microbiome consists of diverse sets of bacteria, fungi, archaea, and viruses (24). Under physiological conditions, 1014 bacteria from 200 to 1500 species are approximated to exist in the colon (2526). Alterations to or loss of intestinal microbiome diversity is related to the aggravation of acute GVHD (2728). In a murine acute GVHD model, distinct microbes in the ileum were highly decreased (e.g., Clostridiales and phylum Firmicutes) or increased (e.g., Lactobacillus johnsonii) compared to bone marrow transplanted control mice without GVHD counterparts. L. johnsonii participated in the amelioration of acute GVHD by suppressing Enterococcus spp. (27). Inhibition of the production of the antimicrobial peptide α-defensin by Paneth cells reduced the physiological diversity of the microflora and permitted expansion of Escherichia coli in GVHD mice (28). Antibiotic treatment to reduce gram-negative bacteria in the GI tract ameliorated acute GVHD severity (29). Shifts in the gut microbiota towards enterobacteria, enterococci, and Bacteroides/Prevotella spp. are associated with increased inflammatory responses in intestinal GVHD (11). Thus, the intestinal microbiota could potentially be manipulated to improve allo-HSCT outcomes.
Innate pattern recognition receptors (PRRs), such as TLRs and nucleotide oligomerization domain (NOD)-like receptors (NLRs), recognize intestinal bacterial pathogens and/or pathogenic molecules. Ligand binding by the TLRs and NLRs expressed on host and/or donor-derived APCs substantially amplifies the release of inflammatory mediators (30). The transfer of HoxB8 neutrophils that lack expression of TLR 2, 3 4, 7, and 9 reduced GVHD severity compared with the transfer of WT HoxB8 neutrophils, indicating that TLR signals promote GVHD development (31). Conditioning-induced GI damage allows the translocation of outer membrane-derived endotoxins from gram-negative bacteria (e.g., lipopolysaccharide (LPS)) into systemic circulation (113233). The binding of LPS to TLR4 accelerated lethal intestinal GVHD by stimulating the production of inflammatory cytokines (e.g., TNFα, IL-1, IL-6, IL-10, IL-12, and TGFβ) from gut-associated lymphoid tissues (GALTs) and macrophages, and IFN-γ from activated donor T cells (934). The endogenous TLR4 agonist heparan sulfate activated dendritic cells (DCs) and aggravated acute GVHD (35). Unexpectedly, however, Tlr4–/– mic developed fulminant GVHD, and allogeneic hosts with a TLR4 mutation (C3H/HeJ mice) had increased intestinal damage compared to wild type counterparts (3637). TLR4 signaling mediated protective effects during GVHD, characterized by reduced intestinal cell apoptosis compared to that in hosts that did not undergo TLR4 signaling (36). In addition, TLR4 ligands were not necessary for the maturation of host APCs for GVHD induction (37). Collectively, these finding suggest that TLR4 signaling is involved in both positive and negative regulation of GVHD. Tlr9–/– mice developed less severe acute GVHD post-HSCT than controls (1138). Consistent with these findings, treatment of wild type mice with a synthetic TLR9 agonist (CpG oligonucleotides) markedly accelerated GVHD severity (39), and treatment with the TLR9-inhibitory oligonucleotide (iODN) 2088 reduced apoptosis of colonic cells in intestinal GVHD (1139). Thus, TLR9 signaling is associated with the induction of intestinal GVHD.
Application of the TLR7/8 agonist R-848 (resiquimod) promoted substantial innate immune activation and T cell migration into target organs (40). Another TLR7/8 agonist, 3M-011, caused differential effects on GVHD depending on the timing of the treatment. Administration of 3M-011 after allogenic transplant increased GVHD mortality, but pre-treatment with 3M-011 reduced the damage to target organs by inducing IDO expression in the colon (394142). Alterations to TLR2 expression on recipient lymphoid and myeloid cells from splenocytes had little effect on acute GVHD (43) (Table I). Thus, each of the TLRs is involved in acute GVHD to a different extent (43444546). Reports on the functional associations of TLRs and their adaptor molecules with GVHD are summarized in Table I and Table II.
MYD88-DEPENDENT EXPANSION OF MYELOID-DERIVED SUPPRESSOR CELLS (MDSCS) IN ACUTE INTESTINAL GVHD
MyD88 is an adaptor molecule that activates inflammatory responses downstream of TLR ligand ligation (Table II) (474849). All TLRs, except TLR3, transduce signals through MyD88 (50). In MyD88-deficient recipient mice, the infiltration of donor T cells into the intestines and the apoptosis of colon cells were reduced, resulting in improved survival and clinical scoring for acute intestinal GVHD (11). However, MyD88-deficiency in donor bone marrow (BM) cells aggravated GVHD, resulting in increased intestinal pathology (51). The exacerbation of intestinal GVHD in recipients of MyD88-deficient BM cells was associated with insufficient expansion of MDSCs from the transplanted MyD88-deficient stem cells. These findings indicate that MyD88 signaling in donor cells promotes MDSC expansion and immune suppression in acute GVHD. The transfer of WT MDSCs into recipients of MyD88-deficient BM cells ameliorated intestinal GVHD, which supports a role for MyD88 in driving MDSC expansion in GVHD. Thus, MyD88 signaling has opposite impacts on intestinal GVHD, depending on whether MyD88 is expressed by host or donor cells.
MDSCs consist of two main subtypes: granulocytic/polymorphonuclear MDSCs and monocytic MDSCs. The phenotypes CD11b+LyG6+Ly6Clow and CD11b+LyG6lowLy6Chigh are used to identify the respective populations in mice. MDSCs expand robustly in various pathological conditions, such as cancers (52), autoimmune diseases (53), inflammation (54), infectious diseases (55565758), and GVHD (49515960). Most MDSC biology has been studied in tumor microenvironments, and preclinical and clinical tumor therapies have been tested for their ability to block MDSC expansion and function. Inhibitors of vascular endothelial growth factor (VEGF; bevacizumab) (61), signal transducer and activator of transcription 3 (STAT3; sunitinib) (62), arginase (NOHA) (52), inducible nitric oxide synthase (iNOS; nitroaspirin) (63), and cyclooxygenase-2 (COX2; celecoxib) (64), as well as agents that induce MDSC apoptosis and necrosis (gemcitabine and IL4Rα aptamer), have been shown to decrease MDSC expansion and tumor growth (6566). The expansion and functional enhancement of MDSCs are required for the control of acute intestinal GVHD. Arginase-1, iNOS, reactive oxygen species (ROS), and nitric oxide (NO) are mediators of the suppressive functions of MDSCs (52). Inflammatory mediators such as COX-2, G-CSF, GM-CSF, IFN-γ, IL-6, IL-10, VEFG, and prostaglandin E2 induce the differentiation and expansion of MDSCs, and inhibit the differentiation of mature myeloid cells in pathogenic environments (6768). These mediators could be targeted to enhance the suppressive functions of MDSCs to ameliorate GVHD. The selective modulation or exploitation of MyD88-mediated signaling to induce MDSC expansion and functional enhancement could be a strategy to suppress acute intestinal GVHD.
CONCLUSION
The dysregulation of microbial homeostasis and TLR signaling-mediated inflammatory responses are involved in the pathogenesis of intestinal GVHD. Understanding the effects and cellular/molecular mechanisms of TLR/MyD88 signaling on innate immune regulation of gut bacteria and MDSCs would aid the development of specific immune modulators to treat intestinal GVHD.


 XML Download
XML Download