

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abbreviations
AIEC
adherent-invasive E. coli
A. parvulum
Atopobium parvulum
B. fragilis
Bacteroides fragilis
B. vulgatus
Bacteroides vulgatus
B. adolescentis
Bifidobacterium adolescentis
B. wadsworthia
Bilophila wadsworthia
CEACAM6
carcinoembryonic antigen-related cell adhesion molecule 6
C. difficile
Clostridium difficile
CDI
Clostridium difficile infection
C. scindens
Clostridium scindens
CD
Crohn's disease
DCs
dendritic cells
DCA
deoxycholic acid
DSS
dextran sulfate sodium
D. invisus
Dialister invisus
E. faecalis
Enterococcus faecalis
ETBF
Enterotoxigenic Bacteroides fragilis
E. coli
Escherichia coli
E. rectale
Eubacterium rectale
F. prausnitzii
Faecalibacterium prausnitzii
FMT
fecal microbiota transfer
FODMAPs
fermentable oligosaccharides, disaccharides, monosaccharides, and polyols
F. nucleatum
Fusobacterium nucleatum
GI
gastrointestinal
H.hepaticus
Helicobacter hepaticus
hGB
humanized gnotobiotic
H2S
hydrogen sulfate
IBD
inflammatory bowel disease
K. pneumoniae
Klebsiella pneumoniae
pks
polyketide synthases
PSA
polysaccharide A
Treg
regulatory T
R. hominis
Roseburia hominis
R. albus
Ruminococcus albus
R. bromii
Ruminococcus bromii
R. callidus
Ruminococcus callidus
SCFAs
short chain fatty acids
UC
ulcerative colitis
INTRODUCTION
In recently years, technological advances in microbiological research, such as next generation sequencing, improved anaerobic culture methods, high-throughput analysis of luminal metabolites, and wider availability of gnotobiotic (GB) mouse models, have revolutionized our understanding of the functional impact gut microbiota has on host physiology and disease. Mounting evidence suggests that the gut microbiota interacts with the host innate and adaptive immune system. For instance, the gut microbiota plays key roles in the maturation and differentiation of Th17 cells, regulatory T (Treg) cells, innate lymphoid cells and immunoglobulin A-producing B cells (123). Additionally, the resident gut microbiota is crucial for the protection of the host against pathogenic microorganisms. The gut microbiota directly inhibits the growth and/or colonization of incoming pathogens by competing for limited nutrients and occupying the potential intestinal habitat (4). Furthermore, the resident gut microbiota protects the host against pathogen colonization through its involvement in mucus production, enhancement of epithelial cell barrier function, the release of antimicrobial peptides, and the production of immunoglobulins (56). Likewise, metabolites generated by the gut microbiota are essential for the maintenance of intestinal homeostasis (7). Short-chain fatty acids (SCFAs), the end products of anaerobic bacterial fermentation of dietary fiber, regulate anti-inflammatory immune responses and enhance epithelial cell barrier function (78). Gut dysbiosis results in the alteration of the intestinal metabolic profile, causing perturbation of gut immune homeostasis (9). Thus, the composition of the gut microbiota is strongly associated with host physiology and perturbations of the gut microbial communities lead to the development of various GI disorders, including inflammatory bowel disease (IBD) (1011). Here, we review the recent advances in the field, focusing on host-microbial cross-talk in IBD.
Go to : 
IBD-ASSOCIATED DYSBIOSIS IN THE GUT
Decreased beneficial bacteria
It has been extensively reported that the abundance of beneficial commensal bacteria in IBD patients, both ulcerative colitis (UC) and Crohn's disease (CD), is reduced. Several studies using meta-genomics analysis have demonstrated that members of the phylum Firmicutes are less abundant in patients with UC or CD (121314). Among Firmicutes, Clostridium clusters XIVa and IV are largely underrepresented in the gut of IBD patients (151617). Clostridium cluster XIVa comprises species belonging to the Clostridium, Ruminococcus, Lachnospira, Roseburia, Eubacterium, Coprococcus, Dorea and Butyrivibrio genera. Clostridium cluster IV consists of Clostridium, Ruminococcus, Eubacterium and Anaerofilum genera. Faecalibacterium prausnitzii (F. prausnitzii), which belongs to Clostridium cluster IV, is well known to be less abundant in ileal biopsy and fecal samples isolated from CD (1415181920) and UC patients compared to healthy subjects (21). Notably, the most significant reduction in the abundance of F. prausnitzii is observed in patients with active IBD rather than those in remission (19) and patients with recurrent IBD (18), suggesting that F. prausnitzii plays anti-inflammatory roles in the gut. Sokol et al. showed that human PBMC stimulation with F. prausnitzii induced the production anti-inflammatory IL-10, whereas it inhibited the secretion of pro-inflammatory cytokines, such as IL-12 and IFN-γ (18). Oral administration of F. prausnitzii in the TNBS-induced colitis model promoted IL-10 production, and reduced IL-12 and tumor necrosis factor (TNF)-α levels in the colon, thereby ameliorating experimental colitis. Furthermore, metabolites produced by F. prausnitzii inhibited NF-κB activation in the mucosa and induced IL-8 production (18). F. prausnitzii can produce butyrate by metabolizing diet-derived polysaccharides and other host-derived substrates (2223). Butyrate is a major source of energy for colonic epithelial cells and is essential for the maintenance of colonic mucosal health. For example, microbiota-produced butyrate induces the development of regulatory T cells (2425) and its administration restores intestinal homeostasis in rats by suppressing IL-17 production in Th17 cells, suggesting butyrate plays a critical role in the regulation of Treg/Th17 balance (26). In addition to F. prausnitzii, accumulating evidence indicates that the abundance of other butyrate-producing bacteria in the human gut, such as Roseburia hominis (R. hominis) and Eubacterium rectale (E. rectale), which belong to phylum Firmicutes, is reduced in IBD patients compared to control subjects (1421). Perhaps not surprisingly, the intestinal levels of the other SCFAs, including acetate and propionate, are also known to be perturbed in IBD patients due to gut dysbiosis. Dialister invisus (D. invisus) is capable of generating both acetate and propionate (27), and the abundance of this bacterium is reduced in patients with CD (15). The most abundant bacterial families represented in the human gut microbiota are Ruminococcaceae and Lachnospiraceae (phylum Firmicutes, class Clostridia). Several studies have reported that the abundance of these families is decreased in IBD patients (23). The abundance of Ruminococcus albus (R. albus), Ruminococcus callidus (R. callidus), and Ruminococcus bromii (R. bromii) in the fecal samples of CD patients was >5-fold lower than in healthy control subjects (14). R. albus produces acetate by degrading dietary cellulose (28). Acetate is the most abundant SCFAs in the colon and the microbiota-produced acetate is utilized by other bacteria, such as F. prausnitzii and Roseburia spp., to generate butyrate (29). While R. callidus and R. bromii can degrade other complex polysaccharides, including starch and xylan, and the degradation products have an impact on the gut microbial community as well as human health (3031), dietary cellulose and polysaccharides are important energy sources for intestinal microorganism (Table I).
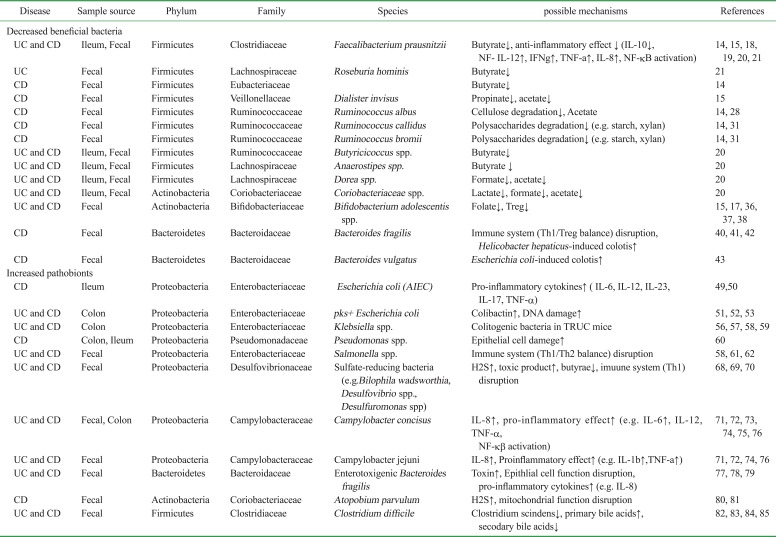
Table I
IBD-associated dysbiosis in the gut
| Disease | Sample source | Phylum | Family | Species | possible mechanisms | References |
|---|---|---|---|---|---|---|
| Decreased beneficial bacteria | ||||||
| UC and CD | Ileum, Fecal | Firmicutes | Clostridiaceae | Faecalibacterium prausnitzii | Butyrate↓, anti-inflammatory effect ↓ (IL-10↓, NF- IL-12↑, IFNg↑, TNF-a↑, IL-8↑, NF-kB activation) | 14, 15, 18, 19, 20, 21 |
| UC | Fecal | Firmicutes | Lachnospiraceae | Roseburia hominis | Butyrate↓ | 21 |
| CD | Fecal | Firmicutes | Eubacteriaceae | Butyrate↓ | 14 | |
| CD | Fecal | Firmicutes | Veillonellaceae | Dialister invisus | Propinate↓, acetate↓ | 15 |
| CD | Fecal | Firmicutes | Ruminococcaceae | Ruminococcus albus | Cellulose degradation↓, Acetate | 14, 28 |
| CD | Fecal | Firmicutes | Ruminococcaceae | Ruminococcus callidus | Polysaccharides degradation↓ (e.g. starch, xylan) | 14, 31 |
| CD | Fecal | Firmicutes | Ruminococcaceae | Ruminococcus bromii | Polysaccharides degradation↓ (e.g. starch, xylan) | 14, 31 |
| UC and CD | Ileum, Fecal | Firmicutes | Ruminococcaceae | Butyricicoccus spp. | Butyrate↓ | 20 |
| UC and CD | Ileum, Fecal | Firmicutes | Lachnospiraceae | Anaerostipes spp. | Butyrate ↓ | 20 |
| UC and CD | Ileum, Fecal | Firmicutes | Lachnospiraceae | Dorea spp. | Formate↓, acetate↓ | 20 |
| UC and CD | Ileum, Fecal | Actinobacteria | Coriobacteriaceae | Coriobacteriaceae spp. | Lactate↓, formate↓, acetate↓ | 20 |
| UC and CD | Fecal | Actinobacteria | Bifidobacteriaceae | Bifidobacterium adolescentis spp. | Folate↓, Treg↓ | 15, 17, 36, 37, 38 |
| CD | Fecal | Bacteroidetes | Bacteroidaceae | Bacteroides fragilis | Immune system (Th1/Treg balance) disruption, Helicobacter hepaticus-induced colotis↑ | 40, 41, 42 |
| CD | Fecal | Bacteroidetes | Bacteroidaceae | Bacteroides vulgatus | Escherichia coli-induced colotis↑ | 43 |
| Increased pathobionts | ||||||
| CD | Ileum | Proteobacteria | Enterobacteriaceae | Escherichia coli (AIEC) | Pro-inflammatory cytokines↑ ( IL-6, IL-12, IL-23, IL-17, TNF-α) | 49, 50 |
| UC and CD | Colon | Proteobacteria | Enterobacteriaceae | pks+ Escherichia coli | Colibactin↑, DNA damage↑ | 51, 52, 53 |
| UC and CD | Colon | Proteobacteria | Enterobacteriaceae | Klebsiella spp. | Colitogenic bacteria in TRUC mice | 56, 57, 58, 59 |
| CD | Colon, Ileum | Proteobacteria | Pseudomonadaceae | Pseudomonas spp. | Epithelial cell damege↑ | 60 |
| UC and CD | Fecal | Proteobacteria | Enterobacteriaceae | Salmonella spp. | Immune system (Th1/Th2 balance) disruption | 58, 61, 62 |
| UC and CD | Fecal | Proteobacteria | Desulfovibrionaceae | Sulfate-reducing bacteria (e.g. Bilophila wadsworthia, Desulfovibrio spp., Desulfuromonas spp) | H2S↑, toxic product↑, butyrae↓, imuune system (Th1) disruption | 68, 69, 70 |
| UC and CD | Fecal, Colon | Proteobacteria | Campylobacteraceae | Campylobacter concisus | IL-8↑, pro-inflammatory effect↑ (e.g. IL-6↑, IL-12, TNF-α, NF-kβ activation) | 71, 72, 73, 74, 75, 76 |
| UC and CD | Fecal | Proteobacteria | Campylobacteraceae | Campylobacter jejuni | IL-8↑, Proinflammatory effect↑ (e.g. IL-1b↑,TNF-a↑) | 71, 72, 74, 76 |
| UC and CD | Fecal | Bacteroidetes | Bacteroidaceae | Enterotoxigenic Bacteroides fragilis | Toxin↑, Epithlial cell function disruption, pro-inflammatory cytokines↑ (e.g. IL-8) | 77, 78, 79 |
| CD | Fecal | Actinobacteria | Coriobacteriaceae | Atopobium parvulum | H2S↑, mitochondrial function disruption | 80, 81 |
| UC and CD | Fecal | Firmicutes | Clostridiaceae | Clostridium difficile | Clostridium scindens↓, primary bile acids↑, | 82, 83, 84, 85 |
![]()
Accumulating evidence has demonstrated that several environmental factors affect the development and progression of GI diseases, including IBD, through the induction of intestinal dysbiosis (3233). For instance, recent meta-analysis results revealed that smoking leads to a decrease of butyrate-producing Anaerostipes spp.. Anaerostipes spp. belong to family Lachnospiraceae and convert lactate to butyrate. The abundance of these bacterial species has been shown to be reduced in IBD patients who smoke (20). Antibiotic use is recognized as the most influential factor linked to disturbances in microbial community structure and function. Some reports suggest that exposure to one or more antibiotics in the first year of life correlates with higher incidence of pediatric IBD (3435). Disturbance of the gut microbiota could be a possible mechanism linking IBD development to antibiotic treatment. Indeed, the abundance of Dorea spp., Butyricicoccus spp., and Coriobacteriaceae spp. is reduced in patients with IBD who have been treated with antibiotics such as ciprofloxacin and metronidazole (20). Furthermore, fecal microbiota analysis of samples isolated from IBD patients has shown an attenuation of Bifidobacterium adolescentis (B. adolescentis), a folate-producing bacterium (151736). Folate (also known as vitamin B9) is involved in several metabolic pathways that are required for cell division. In fact, folate supplementation is recommended for IBD patients due to its ability to regulate rectal cell proliferation (37). Moreover, folate is known to enhance the survival of Foxp3+ regulatory T (Treg) cells, thus diminishing intestinal inflammation through stabilization of the intestinal Treg population (38). These results suggest that organic compound-producing bacteria regulate intestinal homeostasis. In IBD, the abundance of these beneficial bacteria is reduced, thereby resulting in impaired gut function (Table I).
Bacteroides make up the predominant portion of the gut microbiota and play a major role in metabolic activities (39). One member of this genus, Bacteroides fragilis (B. fragilis), has been shown to have a beneficial effect on the host immune system. B. fragilis produces polysaccharide A (PSA), which is recognized as a symbiosis factor used to regulate pathogenic/Treg cell balance. It also exerts anti-inflammatory effects. PSA from B. fragilis is presented by intestinal dendritic cells (DCs) and activates CD4+ T cells, thereby inducing the differentiation of Foxp3+ Tregs. It also induces the secretion of an anti-inflammatory cytokine IL-10, which in turn suppresses the production of pro-inflammatory cytokines, such as IL-17, IL-23, and TNF-α (4041). Additionally, mono-colonization of germ-free mice with PSA-producing B. fragilis modulated pathogenic/regulatory T cell balance and, therefore, protected the host from Helicobacter hepaticus (H.hepaticus)-induced intestinal inflammation (4142). Bacteroides vulgatus (B. vulgatus) is the numerically predominant Bacteroides species in the human gut microbiota. A previous study has shown that B. vulgatus abrogates Escherichia coli
(E. coli)-induced colitis in IL-2 deficient gnotobiotic mice, but the mechanism is unknown (43). Notably, the abundance of B. fragilis and B. vulgatus is lower in fecal samples of CD patients compared to those of healthy subjects (14) (Table I).
Increased pathobionts
As we have reviewed so far, the abundance of beneficial commensal bacteria is significantly decreased in IBD patients. Also, mounting evidence demonstrates that certain members of commensal bacteria, which harbor potential pathogenic features, referred to as pathobionts, are enriched in IBD patients. For example, several microbiome analyses have revealed there is an expansion of the Proteobacteria phylum in IBD patients (121344). The phylum Proteobacteria is composed of six classes: Alphaproteobacteria, Betaproteobacteria, Gammaproteobacteria, Deltaproteobacteria, Epsilonproteobacteria and Zetaproteobacteria. Among these, class Gammaproteobacteria is largest and includes E. coli. Several lines of evidence indicate that certain pathogenic E. coli strains accumulate in the intestinal mucosa/stool of IBD patients, especially CD (14454647). For instance, E. coli strains that display adherent and invasive properties, called adherent-invasive E. coli (AIEC) (48), have repeatedly been isolated from IBD patients. AIEC was isolated from >35% of terminal ileum specimens isolated from CD patients, while only from 6% of healthy samples (49). AIEC can invade intestinal epithelial cells and reach the intestinal barrier, resulting in tissue damage and inflammation. AIEC colonizes the gastrointestinal tract via a type-1 pilus variant that recognizes the carcinoembryonic antigen-related cell adhesion molecule 6 (CEACAM6), which is abnormally expressed by ileal epithelial cells of CD patients (50). In the inflamed ileal mucosa, expanded expression of CEACAM6 augments the intracellular AIEC expansion, thereby leading to a massive production of pro-inflammatory cytokines, such as IL-6, IL-12, IL-23, IL-17, and TNF-α (50) (Table I). In addition to AIEC, polyketide synthase (pks)+
E. coli is known to be pathogenic in IBD (51). It is noteworthy that pks+
E. coli strains are more abundant in patients with IBD than in control subjects. The pks gene is a genomic island of pathogenicity that encodes multi-enzymatic machinery for the production of a bacterial genotoxin called colibactin (52) (Table I). It has also been reported that pks+
E. coli induces DNA damage in vivo (53) and promotes invasive carcinoma in the murine colorectal cancer (CRC) model (51). Given the evidence that IBD is a risk factor for CRC (5455), the more abundant genotoxic bacteria might be responsible for the increased susceptibility to CRC associated with IBD, although the pathogenic contribution of pks+
E. coli to intestinal inflammation per se remains unclear.
Klebsiella spp., Pseudomonas spp., and Salmonella spp. (phylum Proteobacteria, class Gammaproteobacteria) are implicated in IBD (565758). Indeed, when Klebsiella pneumoniae (K. pneumoniae) is used to colonize T-bet Rag deficient ulcerative colitis (TRUC) mice (IBD-prone mouse model) it causes the development of colitis, indicating it is a pathobiont (59) (Table I). Bacterial 16S PCR analysis showed that Pseudomonas spp. were detected in 58% of ileal biopsy samples from pediatric CD patient with CD versus 33% of healthy children (58). Pseudomonas spp. have the ability to attach to intestinal epithelial cells and deliver toxins directly into host cells through a type 3 secretion system, thereby causing epithelial cells damage (60) (Table I). A cohort study has shown that Salmonella infection is associated with increased risk of IBD (58) (Table I). Salmonella infection is recognized as a major health problem worldwide. The infection triggers intestinal colitis by disrupting Th1/Th2 balance (61). Interestingly, Salmonella cannot induce colitis in Nod1 and Nod2-deficient mice (62). This suggests that the sensing of Salmonella is mediated by Nod1 and/or Nod2. Since loss-of-function mutations of the NOD2 gene are associated with increased risk of CD (63), Salmonella infection itself may not trigger inflammation in CD patients who harbor NOD2 mutations. Rather, luminal environmental changes elicited by Salmonella might increase the susceptibility of the host to diseases caused by different pathobionts. Class Deltaproteobacteria (phylum Proteobacteria) includes sulfate-reducing bacteria. The abundance of these bacteria is also known to be increased in patients with IBD (64656667) (Table I). It has been proposed that sulfate-reducing bacteria aggravate gastrointestinal diseases by producing hydrogen sulfate (H2S) and other toxic byproducts as well as by decreasing the availability of beneficial metabolites, such as butyrate (68). Devkota et al. demonstrated that the abundance a sulfate-reducing commensal bacterium Bilophila wadsworthia (B. wadsworthia) was increased due to excessive amounts of taurocholic acid derived from dietary fat. Colonization of IBD-prone IL-10–/– mice by B. wadsworthia resulted in Th1-mediated colonic inflammation (69). Other sulfate-reducing bacteria, such as Desulfovibrio spp. and Desulfuromonas spp., are also more prevalent in patients with IBD compared to healthy individuals (70). The family Campylobacteraceae (phylum Proteobacteria, class Epsilonproteobacteria) is also involved in the pathogenesis of IBD. Recent reports indicate that the abundance of Campylobacter concisus (C. concisus) and Campylobacter jejuni (C. jejuni) in intestinal biopsies or fecal samples isolated from IBD patients is higher than those of non-IBD controls (717273). Others have reported that C. concisus and C. jejuni infections are closely linked to IBD flare-ups (74). For instance, the invasive ratio of C. concisus strains isolated from patients with chronic intestinal disease was >500-fold higher than those from patients with acute intestinal disease or healthy subjects (75). Additionally, attachment of C. concisus and C. jejuni to intestinal epithelial cells prompts IL-8 secretion. The induced IL-8 mediates the recruitment of neutrophils, DCs and macrophages. The recruited cells interact with internalized C. concisus and C. jejuni, resulting in the activation of NF-κB and production of pro-inflammatory cytokines, such as IL-1, IL-6, IL-12 and TNF-α (717276) (Table I). Collectively, several Proteobacteria act as putative pathobionts in IBD and multiple IBD risk factors, including genetics and diet, can induce their uncontrolled expansion.
Enterotoxigenic strains of Bacteroides fragilis (ETBF) produce B. fragilis toxin, which is associated with IBD (77). ETBF was detected in 13.3% of fecal samples isolated from patients with IBD versus 2.9% of control groups. Interestingly, all of the ETBF-positive specimens were isolated from UC and CD patients with active disease (78). Biochemically, the ETBF-produced toxin binds to colonic epithelial cells, inducing the cleavage of E-cadherin, decreasing mucosal barrier function and increasing pro-inflammatory cytokine (IL-8) production (77). Furthermore, ETBF has been shown to cause severe colitis in dextran sodium sulfate (DSS-commonly used to trigger IBD-like experimental colitis in mice) treated wild-type mice, while non-toxigenic B. fragilis did not exacerbate the DSS-induced colitis (79). These findings suggest that the ETBF colonization may contribute to the initiation and aggravation of intestinal colitis, and the enteric microenvironment associated with IBD is conducive to the overgrowth of pathogenic ETBF, resulting in the depletion of symbiotic B. fragilis (Table I). Impaired mitochondrial function is also a defining feature of IBD, especially in pediatric CD (80). Mottawea, W. et al. have recently reported that the levels of one mitochondrial protein, H2S-detoxification protein, were decreased in CD patients. In contrast, the authors observed an increase in the abundance of Atopobium parvulum (A. parvulum) and its abundance positively correlated with disease severity (Table I). A. parvulum is a known producer of H2S. Excessive amounts of H2S in the gut have been shown to cause epithelial cell damage (81). In fact, A. parvulum exacerbated colitis in IL-10–/– mice and the administration of a H2S scavenger ameliorated this phenotype (80). Clostridium difficile (C. difficile) infection (CDI) is a serious complication in IBD patients, because it worsens the long-term course and outcome of disease. Accumulating data suggest that antibiotic-induced dysbiosis associated with IBD can give rise to blooms of C. difficile. In this context, exposure to antibiotics inhibits the conversion of pri mary bile acids to secondary bile acids mediated by com mensal bacteria (82). Since secondary bile acids, such as deoxycholic acid (DCA), can prevent the growth and germination of C. difficile, impaired conversion of primary bile acids to secondary, caused by dysbiosis, weakens colonization resistance of the commensal microbiota against C. difficile (8384). In fact, it has been recently shown that antibiotics decrease the abundance of Clostridium scindens (C. scindens), a bile acid 7 alpha-dehydroxylating bacterium. Decreased abundance of C. scindens can foster the growth of C. difficile due to lower production of DCA (85). Consistently, an imbalance of the microbiome-bile acid pool may lead to increased CDI susceptibility associated with IBD (Table I).
Go to :
THE UTILITY OF A HUMANIZED GNOTOBIOTIC MOUSE SYSTEM
There is a growing body of evidence suggesting that gut dysbiosis contributes to the initiation and pathogenesis of IBD. However, it remains unclear whether IBD-associated gut dysbiosis is related to disease pathogenesis or is merely a secondary change caused by intestinal inflammation and/or medication. To clarify the extent to which dysbiotic microbiotas have a functional impact on disease pathogenesis, recent studies have utilized a gnotobiotic mouse system. In this system, germ-free (GF) mice are colonized with a whole or selected bacterial cocktail isolated from IBD patients. This system, referred to as humanized gnotobiotic (hGB) mouse model or human microbiota-associated mouse (HMA) model, is a powerful tool that can be used to evaluate the functional significance of colonization by putative pathobionts on the host immune system as well as the progression of disease in vivo. The hGB mouse model has been used to prove that mono-colonization with a bacterium or yeast, associated with IBD pathogenesis, impacts host gene expression and metabolic profiles (86). This model has revealed that colonization by Bacteroides thetaiotaomicron markedly facilitates the expression of genes associated with maturation of the immune system, including Treg activation. Additionally, Eun C. S. et al. have shown that a simplified human microbiota consortium, which comprises 7 strains of bacteria that are closely linked to IBD, induces inflammation in IL-10–/– mice through Th1 and Th17 cell responses. This report also demonstrates that colonization by AIEC and Ruminococcus guavus is necessary for the induction of intestinal inflammation, indicating these 2 bacteria play a key role in IBD pathogenesis (87).
In another study, putative IBD pathobionts were rationally selected as “Immunoglobulin A (IgA)-coated” bacteria (88). In both UC and CD patients, subsets of IgA-coated luminal bacteria are enriched compared to healthy control subjects. Strikingly, colonization of GF mice with this IgA-coated bacterial consortium elicits the development of more severe colitis symptoms upon treatment with DSS compared to GF mice colonized with IgA-unbound bacteria. Thus, colitogenic commensal bacteria are enriched in IBD patients and these potential pathobionts are “flagged” by IgA. Since a recent study demonstrated that depletion of IgA-coated bacteria by treatment with an anti-IgA antibody ameliorates colitis (89), therapeutic strategies that target IgA-coated bacteria might be an effective form of treatment for IBD.
In addition to these reports, we have recently used hGB mice to study defective functionality of the IBD-associated microbiota (90). In this study, we utilized a combined multi-OMICS approach to unravel the influence of dysbiosis on the host ecosystem. Structural and functional changes associated with gut dysbiosis were analyzed by 16S rRNA sequencing and functional gene (PICRUSt) analysis, unbiased luminal metabolome analysis, and host gene expression analysis. Furthermore, the impact of IBD-associated dysbiosis on disease pathogenesis was assessed by colonization of GF IL-10–/– mice with IBD and control microbiotas. Based on this study, it is evident that the structural and functional alterations of the gut microbiota present in IBD are, at least to some extent, recapitulated in hGB mice. For instance, α-diversity and richness of the microbiota are significantly reduced in hGB mice colonized with IBD microbiotas compared to hGB mice colonized with healthy microbiotas. These phenotypes are also observed in donor stool samples. Likewise, there are differences between luminal metabolic profiles associated with IBD-hGB mice and healthy-hGB mice. IBD microbiotas differentially impact gene expression in the colon compared to healthy microbiotas. For example, CD patient-derived microbiotas elicit the expression of genes related to pro-inflammatory immune responses, including Th1, Th17, IL-1β pathways. At the same time, CD microbiotas also down-regulate the expression of genes related to the solute carrier group of proteins and the cytochrome families. Notably, the gene expression patterns observed as a result of colonization by CD microbiotas are similar to those occurring in newly diagnosed pediatric CD patients (90). Collectively, this evidence suggests that gut dysbiosis drives changes in intestinal immune responses. Importantly, colonization by CD microbiotas results in the development of severe colitis in GF IL-10–/– mice, an effect not seen with healthy or UC microbiotas. Thus, it is thought that gut dysbiosis in IBD patients is not only a secondary “non-functional” change. Rather, gut dysbiosis has a direct influence on the immune system, making the host more susceptible to disease. Taken together, the gnotobiotic mouse system is a powerful tool that can be used to evaluate the functional role of IBD-associated dysbiosis in the pathogenesis of IBD.
Go to :
DISCUSSION
Several lines of evidence support the notion that gut dysbiosis contributes to the pathogenesis of IBD. As described above, beneficial bacteria are less abundant while pathogenic bacteria are increased in IBD patients (Table I). This imbalance of the gut microbiota is not merely a secondary consequence of disease. Rather, it is thought that a dysbiotic microbiota has inflammatory potential and/or interfere with protective/regulatory function of the immune system. Hence, novel therapeutic strategies that selectively target dysbiosis have attracted considerable interest and offer hope for more effective IBD treatment options. For instance, fecal microbiota transplantation (FMT), which aims to restore normal gut microbial composition and function appears to be an effective treatment for IBD (9192). It has been shown that FMT can be effective for the treatment of CDI in IBD patients, resulting in the normalization of altered microbial composition and metabolic function, and elimination of C. difficile (9394). Indeed, microbial α-diversity and richness in IBD patients treated with FMT increase and become similar to those of healthy donors (95). Additionally, FMT restores secondary bile acid metabolism in the intestine and ameliorates gut damage. Thus, FMT seems to be a promising therapeutic approach that restores/enhances microbiota-mediated resistance to pathogens. Likewise, dietary intervention is another approach used to restore normal microbial composition and function, as it has been reported that diet is a powerful driver of microbial structure in the gut. Elemental diet has been used in the treatment of IBD, in particular to manage the symptoms of CD. Cohort studies have shown that elemental diet has an impact on the composition of the gut microbiota, and induces mucosal healing and clinical remission of CD (9697). Mounting evidence suggests that a diet low in fermentable oligosaccharides, disaccharides, monosaccharides, and polyols (FODMAPs) can effectively reduce GI symptoms in patients with irritable bowel syndrome by altering microbial composition and luminal metabolism (98). In this context, pilot studies have demonstrated that a low-FODMAPs diet can significantly ameliorate gut dysfunction in some IBD patients (9899).
Collectively, recent technological advances have provided evidence that gut dysbiosis is one of the triggers and/or mediators of progression of intestinal inflamma tion in IBD. The increased appreciation of the role the microbiota plays in host physiology and disease has resulted in a vigorous effort to understand the precise mechanism underlying the involvement of the microbiota in the pathogenesis of IBD. Undoubtedly, new discoveries in the field will lead to the development of novel therapeutic approached that aim to restore normal function of the microbiota.
Go to :
 XML Download
XML Download